
摘要:靶向NLRP3炎症小体(inflammasome)是一个爆炸性发展的研究领域。随着这一领域的热情日渐浓厚,几家公司最近开始在临床中测试直接NLRP3抑制剂。同时,NLRP3炎症小体是更大的促炎通路(pro-inflammatory pathway)的一部分,对该通路的探索也正在进行中。已有多个处于临床试验中的分子是针对该通路的多个靶标而产生作用。基于临床前对NLRP3炎症小体积累的机制理解,为评估该领域的当前状况提供了丰富的背景。在此,根据临床和临床前的疗效和安全性数据,我们探索抑制NLRP3炎症小体的尝试。
编译:肖高铿/2020-12-25
原文:Schwaid, A. G.; Spencer, K. B. Strategies for Targeting the NLRP3 In Fl Ammasome in the Clinical and Preclinical Space. J. Med. Chem. 2020, acs.jmedchem.0c01307. https://doi.org/10.1021/acs.jmedchem.0c01307.
前言
靶向NLRP3炎症小体(inflammasome)是一个爆炸性发展的研究领域。不同炎症疾病都会牵扯到炎症小体,在大量文献的推动下,许多制药公司和科技公司将化合物推进到临床试验阶段。包括非酒精性脂肪性肝炎(NASH)、痛风、阿尔茨海默氏症和帕金森氏病等在内的许多适应症都是治疗目标。过去,抑制炎症小体的尝试集中于阻断由炎症小体激活产生的细胞因子。这项工作的结果喜忧参半,商业上的成功也有限。目前,大多数抑制NLRP3炎症小体的尝试集中在直接结合并抑制NLRP3的化合物上。另外,体外和体内研究表明,还有许多其他策略可以干扰NLRP3炎症小体的功能。这就需要在翻译后修饰和蛋白质相互作用的水平上调节NLRP3的细胞机制,以及进一步观察触发炎症小体激活事件上游的策略。几种处于临床研究阶段或已经上市销售的药物碰巧抑制了据称对炎症小体功能至关重要的蛋白。因此,检查其中一些化合物的数据可为NLRP3或其他炎症小体抑制剂的合理临床期望提供依据。最后,基于临床前数据,我们重点关注直接NLRP3抑制剂的安全性,尤其关注抑制NLRP3对感染易感性增加的作用。
NLRP3炎症小体
NLRP3是一种炎症小体传感器蛋白,在许多疾病中都进行过大量的研究。激活NLRP3会产生包括ASC与Caspase-1等在内的寡聚体复合物。该寡聚体蛋白复合物被称为“炎症小体(Inflammasone)”,当复合物中的传感器蛋白为NLRP3时,则该复合物被称为NLRP3炎症小体。通过激活其它炎症小体传感器蛋白可触发炎症小体的形成,其它的传感器蛋白比如AIM2、NLRP1与NLRC4等,这些例子中炎症小体传感器蛋白就是炎症小体复合物的一部分。某些炎症小体传感器可直接识别损伤相关分子模式(damage associated molecular patterns, DAMP)或病原相关分子模式(pathogen associated molecular pattern, PAMP)[1]。比如,NLRP1B识别炭疽致死毒素,NLRC4识别细菌鞭毛蛋白,AIM2识别胞质DNA[2]。另一方面,NLRP3依赖于细胞内第二信使,而不是直接检测外部刺激。 NLRP3的激活可由多种细胞内信号触发。 钾外流是NLRP3炎症小体激活最具代表性的触发因素之一。 NLRP3还对其它细胞内信号应答,例如活性氧(Reactive oxygen species,ROS)和细胞质组织蛋白酶活性[3]。将多种不同的炎症小体触发因素整合到第二信使信号(如钾外流)中,可以解释为什么NLRP3在如此众多的炎症疾病中显得至关重要。炎症小体激活受炎症小体传感器蛋白以外的复杂分子相互作用调控。了解该通路及其分子间相互作用可以勾勒出炎症小体药物开发的其他潜在途径。

图1. NLRP3炎症小体通路以及被临床研究中药物或上市药物抑制的节点
炎症小体的激活由两个关键步骤介导:引发(priming, 通常称为信号1)和激活(通常称为信号2)(图1)。引发包括通过上调NF-κB转录活性来上调与炎症小体相关的蛋白(包括炎症小体传感器蛋白,IL-1β和IL-18)。NF-κB的转录活性受到许多细胞内和细胞外机制的高度调节[4,5]。NF-kB通过IkB在胞浆中处于失活状态。IκB的翻译后修饰或泛素化是对细胞外刺激信号传导的反应,导致细胞核定位和NF-κB的激活。细菌组分通过TLR结合并通过Myd88、IRAK以及TRAF6的信号转导来激活NF-κ B转录活性。诸如IL-1β和TNFα之类的细胞因子也像其他诸如S100a8/a9等之类的DAMP一样激活NF-κB转录活性[6]。在没有NF-κB引发的情况下,许多细胞没有表达足够的炎症小体成分以用于信号2处理后的炎症小体活化 。特别是,许多细胞在不存在信号1的情况下不表达高水平的pro-IL-1β。这会导致在信号2上形成ASC斑点,但不会伴随分泌IL-1β。 已经有许多抑制NF-κB活化策略的报道,但这些策略不在本综述的范围之内[7,8]。
在正常情况下,信号2触发了炎症小体传感蛋白与炎症小体接头蛋白的聚集,并激活Caspase的募集。在NLRP3的情况下,信号2导致NLRP3与ASC的结合与caspase-1募集(图1)。 这种大的蛋白聚集在免疫荧光测定中显示为亮点。Pro-Caspase-1的聚集导致自蛋白水解并产生酶活性的Caspase-1。反过来,Caspase-1切割pro-IL-1β为IL-1β,并且将gasdermin-D切割为N-末端gasdermin-D。 N端gasdermin-D形成膜孔,导致细胞凋亡和IL-1β分泌[9]。
|
|
|
| MCC950 | Dapansutrile |
|---|
图2. 公开结构的两个NLRP3抑制剂
最初,在对人单核细胞分泌的白细胞介素1 β(IL-1 β)用表型筛选二芳基磺酰脲库时发现磺酰脲类化合物格列本脲是一种有效NLRP3抑制剂[10]。随后,进一步优化了这些化合物,并初步研究了它们的机制,初步假设它们通过抑制GST omega 1-111起作用(表1)。 进一步的工作发现这些化合物通过NLRP3抑制起作用。其中一种格列本脲的衍生物CRID3或CP456,773被更名为MCC950,并被发现是NLRP3炎症小体的有效抑制剂[12](图2)。MCC950在类风湿关节炎的临床上进行了测试,但可能由于血清肝酶水平的升高而没有什么进展[13]。MCC950通过与NLRP3的NACHT结构域中的Walker B基序结合并阻断NLRP3介导的ATP水解而发挥作用[14,15]。此后,MCC950被用作一种工具化合物,在一系列涉及NLRP3疾病的体外和体内研究中得到了广泛的应用[12]。这种工具化合物与一系列靶验证技术一起使用,已症明NLRP3在许多疾病中都表现出明显的活性,包括冷冻蛋白相关周期性综合征(cryopyrin associated periodic syndrome,CAPS)、炎症性肠病(IBD)、NASH、痛风、阿尔茨海默病和帕金森氏症[3]。
CAPS包括一系列自身免疫性疾病,包括家族性冷自身炎症综合征(Familial cold autoinflammatory syndrome)和Muckle-Wells综合征[16]。这些罕见的遗传综合征通常由NLRP3基因的功能获得(GOF)突变引起,症状包括发烧、皮疹、关节痛、肌痛、疲劳和结膜炎。 CAPS患者IL-1 β 分泌增加。目前治疗CAPS的药物有IL-1β 阻滞剂canakinumab和anakinra。据推测,直接抑制NLRP3也会减轻这种疾病。支持这一点的是,MCC950在CAPS模型中是有效的,尽管精确的NLRP3 GOF突变似乎对MCC950的疗效很重要[12,17]。虽然CAPS是一种患者群体很小的罕见病,但NLRP3也与更常见的炎性疾病有关,比如如IBD、溃疡性结肠炎和类风湿性关节炎。
在IBD动物模型中,已证明NLRP3激活会导致细胞因子IL-1 β的分泌[18]。虽然这种细胞因子的分泌似乎对IBD的疾病模型有影响,但炎症小体的激活在更大程度可能是通过NOD2驱动的[19]。在NOD2存在的情况下,NLRP3的药理学抑制似乎并不能改善葡聚糖硫酸钠(DSS)模型中的IBD疾病[19]。另一方面,NLRP3在NOD2 -/-动物上DSS诱导的IBD模型中似乎扮演着更重要的角色[19]。虽然NLRP3对IBD的影响可能基于疾病的动物模型尚不清楚,但人类遗传学指出了其更重要的作用。据报道, 携带与NLRP3相互作用的蛋白CARD8 V411I突变的人,其NLRP3活性增加[20]。此外,携带CARD8 V411I突变的患者似乎有望用Canakinumab或anakinra得到治疗[20]。NLRP3的间接抑制也能缓和IBD炎症。例如,已经显示激酶NEK7稳定并激活NLRP3。反过来,在体外和体内敲除NEK7会减弱DSS诱导的WT动物的IBD[21]。
人们也探索通过NLRP3抑制来治疗非酒精性脂肪性肝病(NAFLD)和非酒精性脂肪性肝炎(NASH)。NASH患者的特征是胆固醇和甘油三酯升高[22]。已有证据表明,胆固醇晶体在体外和体内都能激活NLRP3炎症小体[23]。同样,棕榈酸也能激活NLRP3[24]。Loannou等人的证明,以高脂肪饮食饲养的肥胖糖尿病小鼠(一种NASH模型),其肝脂滴富含胆固醇晶体导致库普弗细胞(Kupffer cell)NLRP3的募集和随后的激活[25]。在这种小鼠模型中,他汀类药物和依泽麦布(Ezetimibe)治疗可以阻断胆固醇晶体在脂滴中的积聚,从而改善NLRP3的激活。 MCC950已用于NASH小鼠模型以直接考察NLRP3炎症小体在该疾病中的作用,并减少NAFLD病理和纤维化[2]。
痛风是一种尿酸晶体沉积导致关节炎。治疗痛风的重点是通过别嘌呤醇抑制黄嘌呤氧化酶来阻断尿酸的产生[27]。然而,别嘌呤醇治疗仍然会导致痛风发作。因此,患者同时使用抗炎药(通常是非甾体抗炎药或糖皮质激素)进行治疗[28]。最近,有人假设NLRP3的激活可能对疾病病理学有贡献[29,30]。已证明尿酸晶体在体内和体外可激活炎症小体,而这种激活可被NLRP3 的敲除所阻断。已经采取了好几种策略来阻断尿酸介导的炎症小体活化,包括 β-羟基丁酸治疗,据称是通过阻断炎症小体的组装来阻断IL-β的分泌,在中性粒细胞中咖啡酸阻断ASC的结合来阻断炎症小体的激活[31,32]。NLRP3抑制剂OLT1177,也称为Dapansutrile,在痛风的临床前模型中似乎是有效的, 虽然该化合物的作用机理不如MCC950那般清楚[33,34]。
随着人们对神经炎症在神经退行性病变中的作用认识日益增加,炎症小体在神经科学中的重要性也日益增加。 Gordon等人的研究表明NLRP3和ASC在帕金森病患者中上调,并证明 α-突触核蛋白诱导炎症小体活化,然后从原代啮齿动物小胶质细胞分泌IL-1 β[35]。他们用MCC950和NLRP3敲除的细胞来证明这一过程是由NLRP3利介导的,并将他们的工作扩展到帕金森病的几种体内药效学模型。同样,Heneka等人[36]观察到阿尔茨海默病患者脑裂解物中切割的caspase-1水平升高。通过Caspase-1或NLRP3敲除,携带与家族性阿尔茨海默病相关突变的APP/PS1小鼠免受空间记忆和其他阿尔茨海默病药效学标记的损失。 NLRP3敲除也减少了 β-淀粉样蛋白的沉积。 此外,NLRP3炎症小体激活已被证明是tau病理的驱动因素。NLRP3激活导致tau过度磷酸化和聚集[37]。
这些报告表明了NLRP3抑制剂潜在的多种适应症,其激起人们对NLRP3抑制剂领域的强烈兴奋。NLRP3炎症小体在疾病中的其他作用仍在探索中。炎症和疾病之间的关联数量以及NLRP3对炎症小体激活明确的中心作用,预示着临床上NLRP3炎症小体抑制的机会很多。正如人们所料,这将导致了多家生物技术和制药公司对NLRP3抑制剂研发的巨额投资。当这些努力到达临床时,必将万众瞩目。
表1. 炎症小体抑制剂的结构与靶标
| 结构 | 靶标 |
|---|---|
|
|
Sur1 and NLRP3 |
|
|
NLRP3 |
|
|
GSTO1 inhibitor |
|
|
DUB inhibitor |
|
|
FBXO3 inhibitor |
NLRP3抑制剂专利形势分析
截至2019年11月,共有104份专利申请与潜在的治疗性炎症小体调节剂有关,其中22项专利申请已经授权[38]。专利申请主要集中在NLRP3上,其中IL-1/IL-1R、IL-18、NLRP1和caspase-1受到相当大的关注。 Inflazome和IFM Therapeutics提交的专利申请最多,分别至少10项申请,其次是Onxeo、Nodthera、迈阿密大学和都柏林三一学院,至少有4项专利申请。 自2020年以来,对世界知识产权组织(WIPO)公布的专利申请进行检索发现了另外55份申请。其中,大部分是Novartis、Inflammasome Research Inc.、Inflazome Limited和Native Tumor Immunity Inc申请的。由于申请日和公开日之间有18个月的延迟,预计这个实际申请量还会继续增长。围绕NLRP3专利申请的广度与增长率也说明了投资该领域令人兴奋之处。
临床研究中的直接NLRP3抑制剂
表2. 精选部分临床研究中的NLRP3抑制剂
迄今已有三家公司的NLRP3抑制剂进入临床研究阶段(表2)。Olatec公布了Dapansutrile临床二期用于急性痛风的数据[39]。这是目前临床进展最快的NLRP3抑制剂。在他们的研究中,Dapansutrile在治疗3天时减少50%以上的关节疼痛。Dapansutrile的给药剂量为100、300、1000或2000毫克/天。值得注意的是,超过300毫克/天剂量的疗效没有增加,3天之后的治疗应答在同期组群中相似。在大于等于300mg/天治疗的患者中,第3天的疼痛减轻幅度在56%至68%之间,类似于NSAIDs或强的松龙(prednisolone)观察到的疼痛减轻水平[40]。目前尚不清楚Dapansutrile单药使用是否具有竞争优势。也观测了细胞因子的变化。在300毫克/天或更高剂量给药时,观察到血浆IL-6的剂量变化,在2000毫克/天剂量下,在第一天至第三天期间观察到血浆IL-1β的变化,尽管这一变化不显著。为了评估暴露水平,连续8天给予受试者100、300或1000毫克/天,平均最大稳态血浆浓度分别为36.0、118.6和310.9μM。尽管暴露水平合理,但疗效所需的高剂量意味着NLRP3炎症小体激活的不完全抑制是多药药理的结果。在300μM以上的高剂量和高暴露水平下没有观察到毒性,这也意味着Dapnsutrile的非常有前途。从化学结构上讲,Dapansutrile与其它NLRP3抑制剂有显著的不同。许多专利化合物在结构上类似于NLRP3工具化合物MCC950。然而,Dapansutrile是一种简单的β-磺酰腈(图2)。尽管如此,Marchetti等人的临床前研究表明Dapansutrile抑制了IL-1β的分泌和炎症小体的激活[33]。在重组NLRP3蛋白的研究中,Dapansutrile在1μM或10μM水平降低ATP水解活性。NLRP3炎症小体的选择性研究表明,Dapansutrile并不抑制AIM2或NLRC4炎症小体。
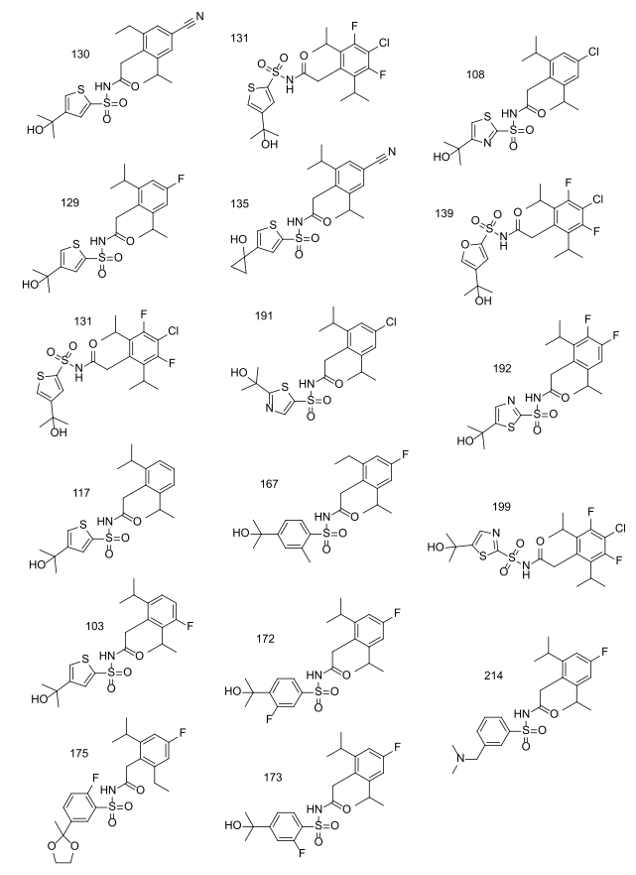
图3. IMF的专利US2019/0119241公开的亚μM活性水平实施例
为了开发NLRP3抑制剂诺华收购了IFM Tre。在收购之前,IFM已经在公开数据库里登记了两项NLRP3抑制剂的一期临床实验,其适应症为冠心病、痛风、NAFLD与克罗恩氏病,并检测作为药效(PD)生物标记物的IL-1β 与IL-18(表2)[41-43]。一项来自IFM的专利披露了化合物结构在某种程度上有别于MCC950(图3)。这些结构保留了磺酰胺但将其中的脲与三环用链接到苯环的乙酰胺进行了替换。实施例化合物的活性为亚μM级别,2,6-烷基取代与4-卤取代是此类化合物的公共特征。另一方面,吡咯环可与一系列不同的芳香杂环(偏好噻吩与噻唑)互相替换[44]。更早期的专利申请披露的结构看起来似乎与MCC950的骨架相似,这意味着临床的化合物有着相似的作用模式。
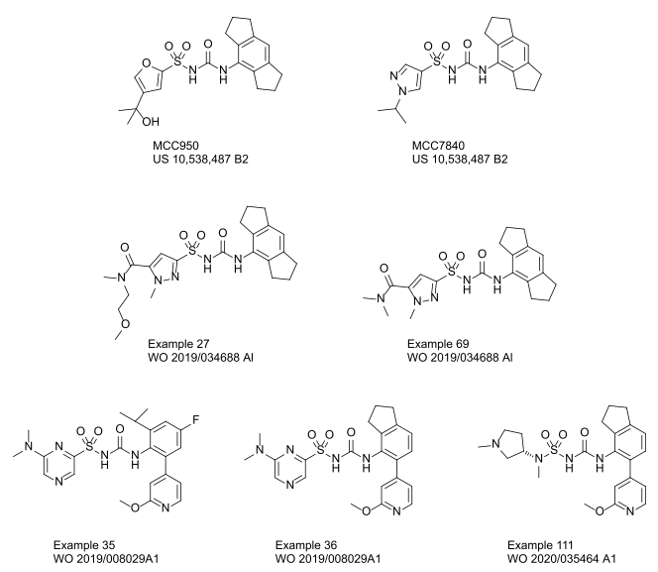
图4. Inflazome专利公开的实施例精选:具有良好的活性与PK
最近被罗氏收购的Inflazome是第2家将NLRP3抑制剂推向临床的公司。这家公司开发了3类化学结构,具有不同的组织分布特征以用于中枢、外周与胃肠道的适应症。Inzomelid是一款中枢透过的NLRP3抑制剂,正在临床1b探索用于阿尔兹海默症、帕金森症、肌萎缩侧索硬化症(ASL),并已经注册了计划开展上述1个或多个适应症的二期临床研究。相反,Inflazome公司的另一个化合物Somalix则被限制为外周的NLRP3抑制剂,其已经完成CAPS的一期临床研究[45-48]。同时,据Inflazome报道,它们还开发了作用于胃肠道的化合物[49]。考虑到NLRP3涉及的适应症非常广,需要一种策略能够狙击某种疾病而不干扰该疾病隔室对疾病没有贡献的NLRP3功能。值得注意的是,IFM也采用了相似的开发策略,它正在寻求具有受限分布特征的化合物。 Inflazome拥有一项授权的专利,它覆盖了MCC950和MCC784050(图4)。Inflazome在2019年公开了24项专利申请,而在2020年截至撰写此综述时,已有6项专利。所有的专利申请都围绕着MCC950的磺酰脲系列探索SAR。在2019年的专利中,他们探索了用极性的亲水取代基替换三环部分以改善PK性质。Inflazome还对噁唑唑进行了替换,探索了各种五元和六元杂环,取代基、烷基与磺酰胺连接[51-54]。本综述精选了专利中具有良好的药效和PK性能的部分实施例(图4)。重要的是要注意,这只是Inflazome在许多实施例合物中的一小部分,具有良好的药效和PK数据,并不是穷尽的列表或最佳化合物列表。此外,无法从这些专利识别出哪个是inzomelid或somalix,因此我们并不知道临床候选药物的结构。
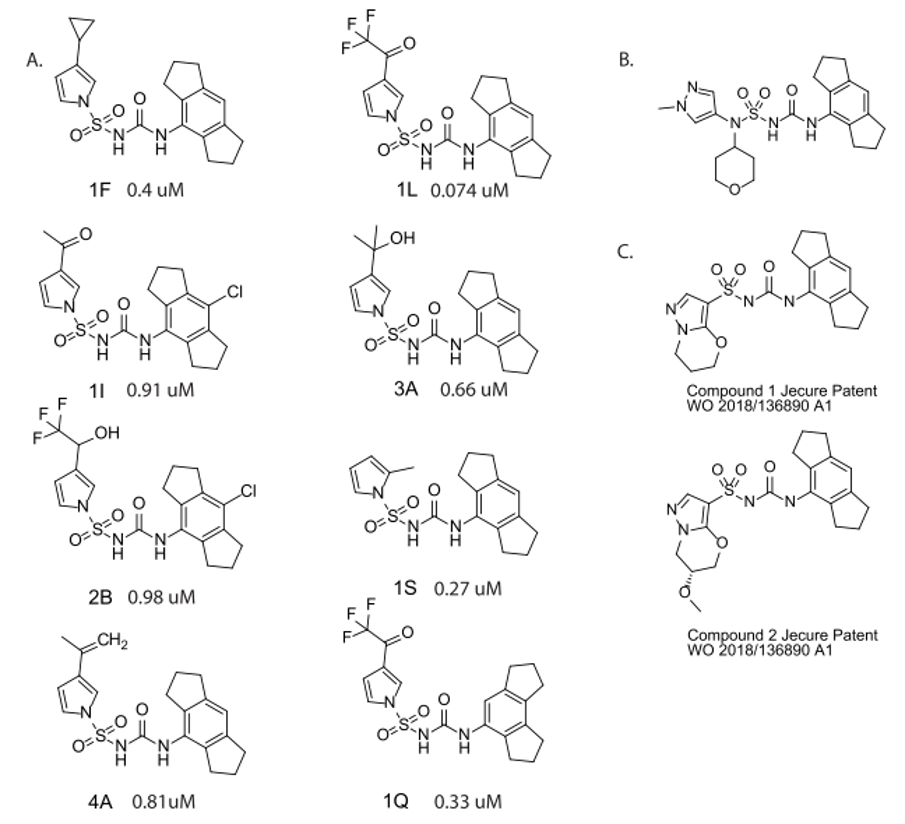
图5. (A)Nodthera的WO2018/015445 A1专利实施例(B)Nodthera的WO2019/121691专利实施例(C)Jecure的WO2018/136890 A1专利实施例
另外两家公司Nodthera和Jecure(已被Genentech收购)也有NLRP3抑制剂处于临床前开发阶段(表2)。其结构尚未公开,也没有对NLRP3炎性小体确切作用模式的公开信息。 Jecure仅拥有一项专利,于2018年公开[55]。该专利围绕MCC950类化学物质,主要围绕5-6个双环吡唑而变化。该专利强调了图5中的Jecure化合物1和2,该化合物在急性体内LPS/ATP应激模型中证明了IL-1β的NLRP3依赖性释放。Nodthera早期专利的结构包含三环氨基甲酸酯/盐结构,但在后来的专利中也包含三环磺酰脲片段(图5)。在他们的WO2018/015445 A1专利中,展示了具有亚微摩尔活性的实施例。这些实例对吡咯进行变化,对MCC950的噁唑进行取代[56]。之后的专利WO2019/121691包含了大量的具有亚微摩尔活性的实施例,这些实施例用各种取代的二唑和脂芳族杂环连接的叔磺酰胺替换了吡咯磺酰胺[57]。还有实施例用嘧啶或异噁唑代替二氮唑。Jecure和Nodthera在专利中要求保护的化合物在结构上与MCC950类似,因此似乎可以通过相似的机理发挥作用[14,55,58,59]。
多家公司将NLRP3抑制剂推到临床凸显了对投资NLRP3作为治疗靶标的认可、并为之兴奋。尚未公开的药物化学差异化策略将随着研发进展而观察到临床差异化。具有相似化学结构、相似NLRP3作用模式的临床差异化尤其令人感兴趣。同时,进入临床研究阶段化合物的数据与靶向炎症小体通路下调炎症小体激活的已批准药物的数据都同样令人感兴趣。
IL-1β抑制剂与Canakinumab
Canakinumab(ACZ885,Ilaris)是由诺华公司开发的人类抗IL-1β单克隆抗体。 Canakinumab是一个信息丰富的临床资产,因为它不仅对所选的适应症成功投放市场,而且还完成了一项大型的III期试验,该试验为IL-1β抑制的有效性和安全性提供了许多见解。我们对Canakinumab的发现和临床进展进行了全面综述[60]。该抗体通过与IL-1β结合并抑制IL-1β的作用而起效,IL-1β通常通过与IL-1R结合而引发炎症。CANTOS试验是Canakinumab作为抗炎疗法的关键性III期试验。该结果于2017年发表[61],描述了对动脉粥样硬化的疗效,安全性以及对患有慢性炎症人群的其他症状和合并症的疗效。该试验的规模为10061名患者、以及随机双盲安慰剂对照、为期48个月的治疗评估,包括对复发性心血管事件、症状和副作用的评估,从而有力地进行了观察以支持研究的结果。
CANTOS试验提供了临床概念验证:即抑制炎症可以直接影响动脉粥样硬化血栓形成患者的反复心血管事件发生率。Canakinumab不降低血脂,其疗效完全来自抗炎机制。虽然不是登记的终点,但观察到治疗组的癌症死亡率显著降低。具体而言,一项事后分析显示,在300毫克组中,肺癌和肺癌死亡的发生率显著降低[60]。虽然治疗组的总死亡人数与安慰剂组相似,但治疗组因感染和败血症死亡的人数显著增加。在3344名安慰剂组患者中,1.8%的患者经历了致命的感染或败血症;而在最高剂量(300mg)组中,Canakinumab治疗的发生率为3.4%[61]。其他非致命的不良事件包括嗜中性粒细胞减少症(可导致感染易感性增加)和血小板减少症,尽管在出血方面没有显著差异。
针对许多罕见病适应症的小型试验中获得的疗效和安全性信息支持了Canakinumab作为抗炎疗法的用途。2009年,Canakinumab被批准用于治疗成人和4岁及以上儿童的CAPS治疗,包括家族性冷自身炎综合征和Muckle-Wells综合征。这项批准是基于Lachmann等人发表的一项临床试验。在这项试验中,每8周使用一次Canakinumab与CAPS儿童和成人的炎症性疾病的实际控制有关[62]。临床终点合并为这些患者通常经历的所有症状的“发作”。对治疗的完全反应被定义为全身性没有疾病活动或最小疾病活动。虽然本试验的样本量很小,只有35名患者,但在一年的时间里,疗效非常显著,因此该药物被批准用于CAPS患者的治疗。
与CANTOS试验一致,在CAPS试验中观察到显著的感染增加。然而,患者人数很少以至于很难解释其统计学显著性。例如,有一名患者因复发性尿路感染而停止Canakinumab治疗。35人中有1人不足以引起关注,但即使是这样一个小规模的研究,感染风险仍然值得注意。重要的是,没有发现死亡或危及生命的不良事件(AE)。批准的使用说明书包括与严重感染发病率增加有关的警告[63]。
在广泛的症状和疾病中观察到的Canakinumab有效性给人们提供了足够的信心,即通过IL-1 β 轴减少全身炎症有可能治疗许多常见疾病,包括心血管事件。与此相关的感染风险可能会阻碍这一领域的进展。 由于已知多种炎症小体可产生IL-1 β,所以有人推测,直接抑制NLRP3介导的炎症小体应答而不是IL-1 β,将会降低严重感染的风险。
双醋瑞因(Diacerein)是一种小分子,据报道可以降低IL-1 β而起效。双醋瑞因是一种蒽醌衍生物,是一种被批准用于骨关节炎的市售药物[64]。其作用机制通常归因于阻断IL-1 β的释放,最近被认为是NLRP3抑制。对临床研究、荟萃分析和上市后监测的文献进行分析,研究了双醋瑞因在膝关节和髋关节骨关节炎对症治疗中的疗效和安全性[64]。结果证实了其对非甾体抗炎药和扑热息痛为禁忌症患者的安全性。双醋瑞因的副作用包括常见的诸如腹泻之类的胃肠道失调、常见的轻微皮肤反应以及罕见的肝胆疾病。严重的感染风险没有被强调,这说明了抑制IL-1 β的安全性和有效性的潜力。最近,TWI Biotechnology已经注册了AC-203,一种1%双醋瑞因的局部用药制剂,用于单纯大疱性表皮松解症(EBS)的临床试验[65]。EBS是一种导致皮肤非常脆弱和容易起泡的遗传疾病。该公司声称,双醋瑞因的作用机制是通过抑制NLRP3炎症小体的激活。TWI Biotechnology公开的唯一数据是一份专利,数据表明前药双醋瑞因(Diacerain)的活性成分大黄酸(rhein)可以抑制ASC和NLRP3的表达,荧光共聚焦显微镜读数法实验表明大黄酸在THP-1细胞中可防止PMA预处理和MSU刺激形成NLRP3炎症小体复合物[66]。
IL-1R拮抗与ANAKINRA
除了Canakinumab之外,阻断IL-β 信号传导的另一方法是IL-1R拮抗。Anakinra是重组的内源性IL-1R拮抗剂蛋白IL-1RA。Anakinra通过IL-1R阻断IL-1α和IL-1β的信号传导。 它也在临床上得到了广泛的探索,目前是一种批准的、尽管不常见的处方药物。
2001年,阿那金拉(Anakinra)被FDA批准用于治疗中、重度类风湿性关节炎,这些患者至少有一种疾病治疗抗风湿药物治疗失败。 然而,它在这一适应症中仅有中等疗效[67]。在使用阿纳金治疗的患者中,38%达到ACR20,而安慰剂治疗的患者为23%。 ACR20是一种应答,定义为关节压痛和肿胀数改善20%、而五个变量中有三个的改善水平相同:患者/医生整体评估、疼痛评分、健康评估问卷评分和实验室急性反应物。然而,Anakinra在RA中的疗效不如抗TNF α疗法有效,而且通常不用于该适应症[68]。Anakinra在CAPS中也有效[69]。尽管它在临床上用于该适应症,但其4-6小时的短半衰期和注射部位反应的频率使得人们更倾向于使用半衰期为26天的Canakinumab[69−71]。
总的来说,阿纳金拉是安全的。然而,虽然总体感染率没有变化,但是与安慰剂相比,在阿纳金拉治疗的患者中观察到严重感染率从0.6%增加到1.8%,总体感染没有增加3倍。肺炎是阿纳金拉治疗组观察到的最常见严重感染[67]。值得注意的是,这种增加在并不具备统计学意义上的显著性,而且由于阿纳拉金临床应用有限,在批准后进一步探索安全性的发现受到阻碍。在临床前动物中,阿纳拉金与依那西普(enteracept,一种TNF α 阻断剂)联合治疗在RA模型中表现为相加或协同作用[72]。然而,在临床试验中没有观察到此类疗效。阿那金拉和依那西普的联合治疗确实增加了严重感染的数量,25mg依那西普QW或BIW与100mg/天的阿那金拉使得严重感染的发病率提高到3.7%或7.4%。
由于IL-1 β 的分泌是炎症小体活化的直接产物,Canakinumab和Anakinra的研究表明,在某些适应症中,直接阻断炎症小体活化的抑制剂可能是有效的。值得注意的是,来自多种不同的炎症小体传感器蛋白的炎症小体活化都对IL-1 β的分泌有贡献。仅靶向单个炎症小体传感器蛋白可改善用抗IL-1 β治疗观察到的一些安全性信号。
Larock等人回顾了Canakinumab和Anakinra的上市后监测,发现总体感染率较低[73]。低感染风险的例外是A组链球菌(Group A Streptococcus ,GAS)引起的严重侵袭性感染。对于接受IL-1 β抑制剂的患者,感染率显著增加了330倍。根据人和小鼠(IL-1R −/− 敲除小鼠和化合物治疗)的数据,IL-1R拮抗剂阿纳金拉与IL-1 β信号的减少相关,并使得GAS生长和传播增加。作者继续假设,阻断内源性IL-1 β的成熟可能不会带来与阻断受体信号传导相同的GAS感染风险。
最近,NLRP3炎症小体在新冠肺炎诱导的急性呼吸窘迫综合征(ARDS)中的作用也受到了人们的关注[74]。 SARS-CoV-2蛋白orf3a、orf8a和E蛋白可介导钾离子和钙离子的流动, 这介导了NLRP3炎症小体的激活[74]。为了治疗COVID-19诱导的ARDS,对同期组群的29名患者进行了高剂量阿纳金拉治疗[75]。研究人员指出,阿纳金拉的半衰期较短,这使得它可以在需要时进行仔细的监测和停用。与回顾性数据相比,阿纳金拉治疗降低了C反应蛋白,提高了21天的总生存率(90%对56%)[75]。然而,机械通气的时间没有差异,这使得结果并不那么令人振奋。在思考这一数据时,必须牢记的是:研究规模小、单一研究中心和分析的回顾性性质[75]。在进行这项分析时,报道的死亡率有着巨大的差异(不同的报道为28-78%);以及追踪其他靶向炎性细胞因子的抗体治疗新冠肺炎诱导的ARDS的复杂临床数据,在此基础之上的回顾性研究表明:尚需进一步的研究然后才能得出强有力的结论[76-79]。
Caspase-1抑制剂临床势态与结果
抑制caspase-1是阻断炎症小体激活下游炎症小体信号的另一条途径。Pro-caspase-1是炎症小体复合物的关键成分,当被激活为caspase-1时,半胱氨酸蛋白酶将原白细胞介素(pro-IL-1β)和原IL-18(pro-IL-18)切割成其活性细胞因子,是产生IL-1β和IL-18的最后一步[80]。此外,caspase-1会导致Gasdermin-D裂解、膜孔形成和细胞死亡(pyroptosis)[9]。在缺乏caspase-1活性的情况下,临床前研究表明,炎症小体介导的细胞死亡可以通过caspase-8发生[81]。小分子和肽拟似物类Caspase-1抑制剂是一组有前途的临床化合物,目前尚未交付上市药物。 试图选择性地抑制Caspase-1或抑制泛Caspase活性,但尚未有报道一种化合物能够在临床上显示出足够的疗效和安全性。
表3. 靶向NLRP3炎症小体通路的临床化合物
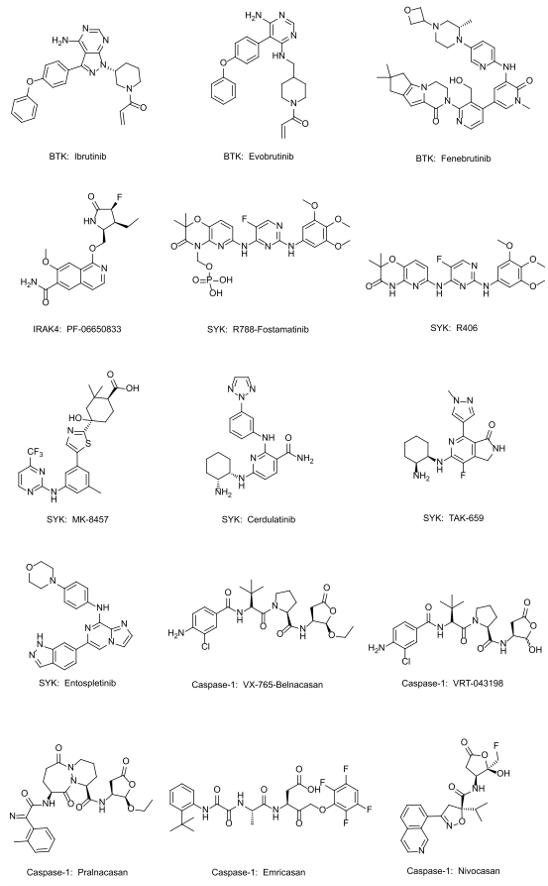
我们重点关注Caspase-1抑制剂VX-765(belnacasan)和VX-740(pralnacasan),它们是临床上进展最快的、最具选择性的Caspase-1抑制剂(表3)。VX-740是由Aventis和Vertex共同开发。VX-740是一种前药,转化为活性代谢物VRT-18858抑制caspase-1[82]。虽然没有人的PK数据发表,但临床前研究表明,转化的VRT-18858在药效学活性和治疗相关范围内[83]。2003年11月,Aventis和Vertex宣布,由于一项动物毒性研究的结果,他们自愿取消了VX-740类风湿关节炎的IIb期临床试验。该研究显示,在高剂量9个月的VX-740暴露后,肝脏出现异常。他们指出,在人体试验中没有发现类似的肝毒性[83,84]。此后,没有开展进一步的VX-740临床试验。
VX-765在结构上与VX-740相似,也是一种前药,它被血浆酯酶转化为活性代谢物VRT-043198。Vertex发现并开发了它的免疫适应症(未披露)、癫痫和银屑病。VRT-043198通过对Caspase-1活性位点中催化性半胱氨酸残基的共价修饰而起作用。与其他半胱氨酸蛋白酶相比,VRT-043198对Caspase-1的选择性很高,只有Caspase-4在相似效力下被抑制[85]。临床前,VX-765在炎症、关节炎和皮肤病的动物模型中显示出有效性[86]。二期临床试验测试了VX-765用于银屑病和癫痫适应症,尽管没有RA。2011年3月,Vertex报告了癫痫二期试验的数据[87]。VX-765是安全的,并进入了IIb阶段;然而,在2012年停止。这些数据表明,用VX-740观察到的初始毒性结果是化合物相关的,而与机制无关。 VX-765为什么没有临床疗效尚不清楚。
进入临床的其他Caspase抑制剂都是广谱Caspase抑制剂,包括IDN-6556(emricasan)和GS-9450(nivocasan)[86]。由于这些化合物是非特异性的,目前尚不清楚观察到的任何疗效或安全性是否与抑制炎症小体信号相关。
抑制NLRP3炎症小体:不只是直接结合
NLRP3通路的上游和下游提供了抑制NLRP3功能的其它机会。一系列蛋白通过直接结合、翻译后修饰或炎症小体蛋白的降解来调节炎症小体的激活。NEK7是一个潜在的具有临床意义的蛋白。利用基于质谱的下拉实验,发现在LPS或ATP激活炎症小体后,NEK7直接与NLRP3结合[88]。已经发现NLRP3和NEK7之间的相互作用是由NEK7催化结构域介导;然而,干扰NEK7催化活性的突变并不影响蛋白-蛋白相互作用。此外,NEK7是鼠骨髓衍生的巨噬细胞(BMDM)中NLRP3炎症小体活化、形成ASC斑点所必需的。包括ATP、金霉素、短杆菌肽、MSU、L-亮氨酸-L-亮氨酸甲酯(LLOMe)、二氧化硅、明矾和焦磷酸钙沉积(CPPD)等在内的许多刺激的实验中观察到了这一关键作用。另外,还表明NLRP3需要NEK7来响应ROS[89]。即使是功能突变体R258W的组成型活性NLRP3也需要NEK7来激活炎症小体。NEK7似乎与NLRP3的多个拷贝结合,这使得NLRP3寡聚物能够桥接[90]。值得注意的是,NEK7有另一个相互作用的伙伴,NEK9,它参与有丝分裂。由于一些细胞表达的NEK7数量有限,因此这些细胞中的NLRP3炎症小体活化只能在间期发生。综上所述,这些数据提出了NEK7及其与NLRP3的相互作用是否是潜在的药理学靶点。
据报道,天然产物冬凌草甲素(Oridonin)通过 α,β-不饱和羰基(Michael受体)共价结合NLRP3 NACHT结构域中的CYS279[91](表1)。与许多报道的NLRP3抑制剂不同,冬凌草甲素阻断NLRP3与NEK7的结合而不是阻断NLRP3 ATP酶活性。在阻断与NEK7的结合中,BMDMs中的NLRP3炎症小体被ATP、尼吉霉素和MSU的激活也被抑制。 支持这一作用机制的是,CYS279突变为丙氨酸阻止了冬凌草甲素介导的炎症小体激活的抑制。
NEK7的翻译后修饰也可以被操纵来阻断NLRP3炎症小体的激活[92]。谷胱甘肽转移酶GSTO1从NEK7中去除谷胱甘肽。这是NEK7介导NLRP3炎症小体激活所必需的。发现氯乙酰胺GSTO1抑制剂C1−27阻断NEK7脱谷胱甘肽化,进而阻断NLRP3炎症小体在体外和体内的激活(表1)。有趣的是,C1-27似乎比LPS/nigericin更有效地阻断LPS/ATP激活NLRP3。
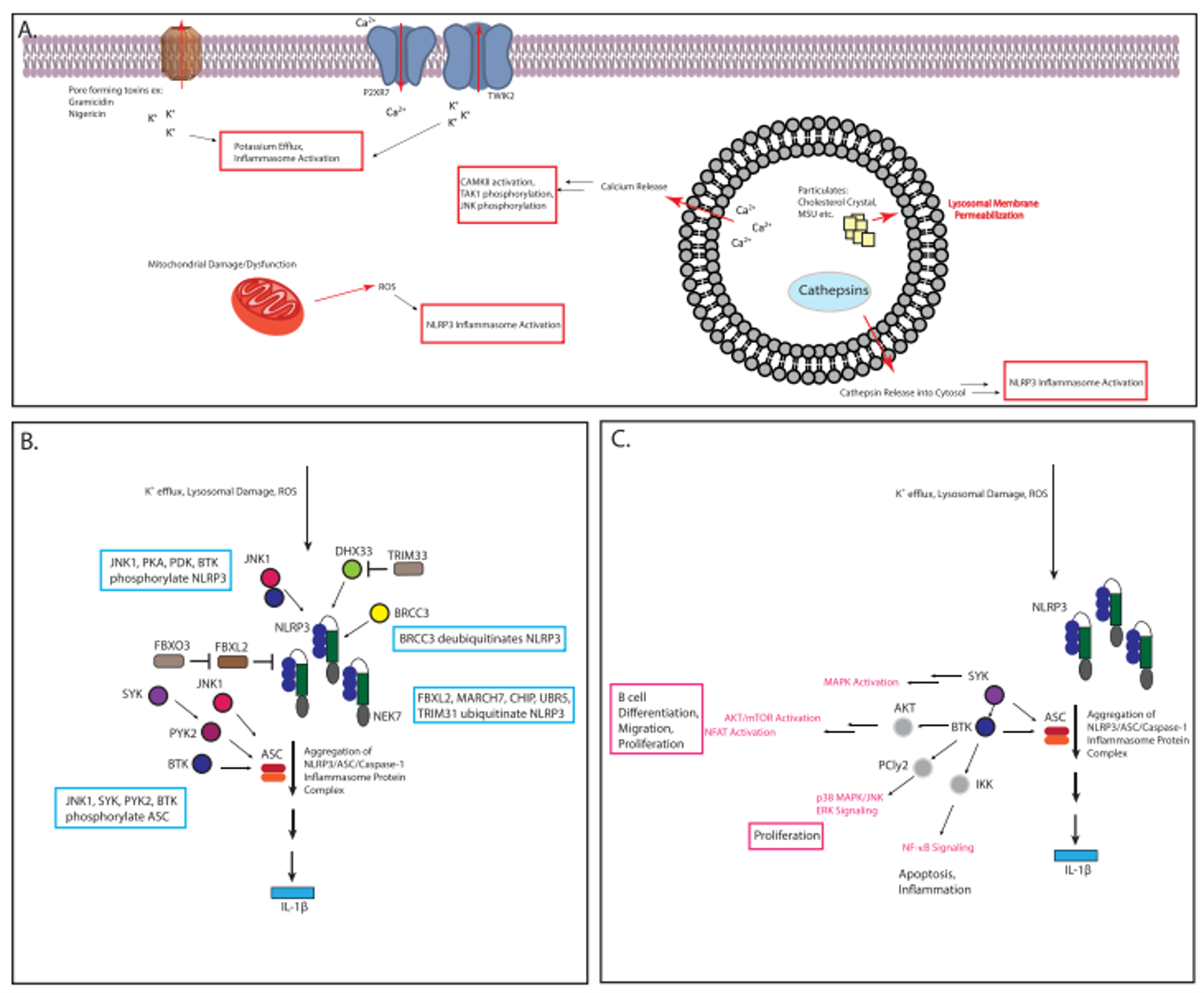
图6. NLRP3激活的调控通路 (A) NLRP3炎症小体激活路径; (B) 调控NLRP3炎症小体成分的Proximal蛋白 (C) 将Syk与BTK整合到炎症小体激活过程及其下游过程
另一个翻译后修饰(post-translational modification, PTM)控制NLRP3的活性是磷酸化[93]。已经证明丝氨酸被JNK1磷酸化是炎症小体活化所必需的。PKD的磷酸化调节NLRP3的亚细胞定位,NLRP3的PKA磷酸化可以阻断其ATP酶活性,从而导致炎症小体抑制[94−97]。NLRP3丝氨酸5处的磷酸化也可以阻断NLRP3和ASC的结合,PP2A的去磷酸化被证明能够实现聚集。同样,ASC的磷酸化对炎症小体的活化也很重要[98]。推测ASC含有几个重要等磷酸化位点。已证明酪氨酸146(在小鼠中为酪氨酸144)对ASC的寡聚化是重要的。激酶JNK1、Syk、Pyk2和BTK与酪氨酸146的磷酸化有关,尽管很难确定这种磷酸化是直接还是下游的(图6)[99]。BTK与ASC和NLRP3形成蛋白质-蛋白质相互作用,增加了BTK介导的磷酸化是直接事件的可能性[100]。BTK还被证明可以磷酸化NLRP3[101]。在体外磷酸化分析中,Pyk2直接磷酸化ASC。这些节点中的每一个都是调节NLRP3功能的潜在成药性节点。
泛素-蛋白酶体系统被认为是一种可成药的蛋白网络[102]。有几种药物通过操纵或抑制泛素连接酶或蛋白酶体来起作用[103]。炎症小体也受泛素连接酶网络的调节(图6)。Py等人筛选出阻断IL-1 β分泌的化合物,发现了阻断NLRP3炎症小体激活的化合物G5[104](表1)。他们确定G5可致K63和K48多泛素聚集。最终,该化合物的功能是阻断BRCC3对NLRP3的去泛素化。在实验的过程中,用免疫共沉淀来识别什么样的去泛素酶(deubiquitinases, DUBS)可与NLRP3相互作用,然后发现了43个DUB。 其中12个在过表达时对NLRP3有去泛素化的作用。FBXL2是一种E3泛素连接酶,它使NLRP3的LYS689泛素化。FBXL2本身受另一种泛素连接酶FBXO3调节。因此,NLRP3炎症小体的活化同时受到FBXL2(降低NLRP3水平)和FBXO3(降低FBXL2水平导致NLRP3水平增加)的翻译后调节。FBXO3小分子抑制剂,如BC-1215,可阻止FBXL2的降解,导致NLRP3的降解增加[105](表1)。已有一期临床试验正在IBD中测试此类分子[106]。其他研究表明,多巴胺可以通过MARCH7介导的泛素化和降解来防止NLRP3炎症小体活化[107]。值得注意的是,这些研究还发现了另外两种能够与NLRP3、CHIP和UBR5免疫沉淀的E3。 另外,还发现泛素连接酶TRIM31导致NLRP3中K48的泛素化以及随后的蛋白酶体降解[108]。有趣的是,LPS和IL-1 β 也上调了TRIM31的表达,尽管上调的动力学比NLRP3慢,这表明TRIM31可能是细胞终止炎症小体信号的一种机制。
除了直接调控NLRP3外,几种已知的相互作用蛋白也受到泛素化的调控。DHX33是一种胞浆dsRNA传感器,可以与NLRP3相互作用来触发炎症小体的激活[109]。这种蛋白质− 蛋白质的相互作用依赖于TRIM的泛素化[33]。此外,ASC丰度和炎症小体传感器的相互作用可受到泛素化的正和负影响[110-112]。与NLRP3结合的其他去泛素化和泛素化酶的存在提出了一个问题,即是否有其它的泛素介导的轴来控制NLRP3的功能。炎症小体本身的上调亦可导致炎症小体的活化,该过程也可以作为抑制的靶标。导致NLRP3炎症小体激活的主要途径有三种:溶酶体膜通透性、ROS和钾外排(图6)[3]。这些过程是相互联系的,因此很难准确地找出激活炎症小体的关键启动过程。然而,大量的文献都集中在解决这个问题上。
溶酶体膜通透性似乎主要是由微粒刺激引起的。有证据表明,MSU、胆固醇晶体、二氧化硅、明矾和LLOMe都是通过诱导溶酶体损伤而起作用的[113]。反过来,假设溶酶体膜通透性通过两种主要途径激活NLRP3炎症介质(图6)。第一种途径是组织蛋白酶释放到胞浆中[23,114]。组织蛋白酶抑制剂似乎可以阻断胆固醇和其他微粒的炎症小体活化。人们通常假设这是通过抑制组织蛋白酶而发生的[113,115]。然而,由于组织蛋白酶B抑制剂的泛作用以及发现其他组织蛋白酶的过表达通常可以在基因上补偿组织蛋白酶敲除,因此很难确定这一点。目前尚不清楚是什么底物或骨架功能组织的蛋白酶在胞浆中激活炎症小体。 溶酶体膜通透性的第二种机制是通过激活上游激酶来激活炎症小体[116]。溶酶体钙流可激活CAMKII,这导致TAK1的磷酸化以及随后的JNK的磷酸化,其如上所述活化炎症小体。CAMKII通路的激活已被证明是由诸如MUS之类的刺激(而不是ATP等其它刺激)驱动的。同样,组织蛋白酶抑制剂似乎对微粒刺激有更大的影响,尽管一些刺激的结果,如尼吉霉素,在文献中是混合的。这种差异被假定为非选择性组织蛋白酶抑制剂的不同多药理学的结果。除了组织蛋白酶抑制剂外,CAMKII抑制剂和吞噬作用抑制剂也被用于减轻溶酶体膜通透性诱导的损伤[23]。此外,还正在研究减少溶酶体破裂的其他方法。例如,二氢硫辛酸通过上调Lamp1和减少通过CAMKII的信号传导来抑制大鼠蛛网膜下腔出血时炎症小体的活化[117]。作为工具,氯化铵和氯喹已被用作溶酶体中和剂,它们似乎通过抑制组织蛋白酶活性来阻断炎症小体活化[118,119]。尽管溶酶体渗透是导致炎症小体活化的一种途径,但它与炎症小体活化的其他机制是相互联系的。例如,ROS的产生会导致随后的溶酶体通透性,而在某些情况下,阻断钾流也会减弱溶酶体通透性引起的炎症小体活化[120]。
研究表明,线粒体毒素可使ROS增加以及随后的NLRP3炎症小体活化[121,122]。有丝分裂抑制剂3-methylade-nine也会导致线粒体损伤、ROS增加和炎症小体的激活。在巨噬细胞和小胶质细胞中也进行了类似的实验。同样,自噬蛋白Beclin和LC3B的敲除会导致线粒体功能失调、ROS和线粒体DNA的释放以及炎症小体的激活[123]。VDAC1或VDAC2的敲低或者用丝裂霉素、线粒体靶向抗氧化剂进行治疗,可阻断ROS诱导的炎症小体激活[121,122]。反过来,线粒体在炎症小体引发前通过P2X7去极化会导致引发后炎症小体的缺乏。假设这是通过HIF-1 α的上调介导的[124]。钾外流是NLRP3炎症小体激活的第三种途径。钾外流似乎是炎症小体活化的一个常见的潜在触发因素,被许多不同的炎症小体激活剂可诱发生成[125]。事实上,在文献中常用作NLRP3炎症激活剂的微生物毒素短杆菌肽和金霉素在细胞膜上形成了钾渗透孔。向细胞培养基中添加高浓度的钾能够阻断许多不同来源的炎症小体活化,这支持钾外流在炎症小体活化中起到关键的作用[126]。参与炎症小体激活的确切钾通道尚不清楚。P2RX7和TWIK-2一直被认为是NLRP3激活的重要钾通道[127]。TWIK-2的敲除或敲低已被证明能阻断ATP引起的NLRP3炎症小体激活,但不能阻断黑曲霉毒素引起的NLRP3炎症小体激活。奎宁已用于药理学阻断TWIK-2并获得类似的结果;然而,必须根据奎宁的多药理学特点仔细地考虑数据。该数据支持将直接抑制钾外流作为一种抑制炎症小体活化途径的可能性。
NLRP3炎症小体受到许多过程调控,包括转录、PTM、PPI以及包括LMP、ROS生成和钾外排在内的上游过程。在细胞和动物模型中,药理学意义地靶向这些过程中可以有效地抑制了NLRP3炎症小体的活化,有时可特异性地抑制单个信号2活化剂。这些过程支持了在临床中用不直接结合NLRP3的分子来抑制NLRP3炎症小体活化的可能性。与直接NLRP3抑制剂相比,这些途径有哪些优点或缺点,还有待观察。然而,临床上的一些抗炎药似乎对上述通路确实有影响。当考虑直接NLRP3抑制剂的基本原理时,综合评价这些化合物的特征是非常有价值的。
非直接NLRP3抑制剂的临床效果
以上所述的导致NLRP3炎症小体活性消融的一些机制已在临床上进行了探索,尽管目的不是抑制炎症小体(图1)。因此,探索这些化合物的临床数据,着眼于炎症小体的抑制,有助于揭示炎症小体通路的作用机制并产出成果。
BTK抑制剂主要用于治疗慢性淋巴细胞白血病(chronic lymphocyte leukemia, CLL),尽管最近BTK抑制剂正在探索治疗自身免疫性疾病[128]。BTK对B细胞存活很重要,对CLL存活很重要的B细胞受体下游的几个通路的激活也是至关重要的[129,130]。如前所述,BTK还通过NLRP3和ASC两者的磷酸化来激活NLRP3炎症小体。这提出了BTK抑制剂是否阻断患者的NLRP3激活以及该抑制在临床上具有什么效果的问题。Liu等人观察到服用不可逆BTK抑制剂依鲁替尼(ibrutinib)的患者在体外刺激后外周血单核细胞(PBMC)分泌的IL-1 β 降低,这表明在体内依鲁替尼对炎症小体有作用[131]。同样,X-连锁无球蛋白血症(X-linked agammaglobulinemia ,XLA)是一种BTK基因发生突变的疾病,该患者的PBMC在体外刺激后表现出较低的炎症小体活化。Evobrutinib是另一种结构相似的不可逆BTK抑制剂,最近在多发性硬化症中的二期临床中获得阳性数据,并且显示出钆增的病变(gadolinium-enhancing lesions)数量减少[132]。Fenebrutinib是一种选择性可逆BTK抑制剂,在类风湿性关节炎中的二期临床中显示了一些阳性数据。在对甲氨蝶呤单独反应不充分的患者中,Fenebrutinib治疗提高了ACR20评分[133](表3)。与TNF-α拮抗剂抗体阿达木单抗(Humira)的护理标准相比,两者的效果相当。假设治疗效果来自对B细胞和髓系细胞的混合效应。需要记住的是,抑制BTK将影响炎症小体之外的许多细胞过程,例如增殖、存活、迁移、血管生成和细胞因子产生,将BTK抑制剂的效力仅归因于减少炎症小体的活化是不准确的。虽然有些疗效可能来自于炎症小体的抑制,但BTK抑制剂的多效作用使我们很难得出完全如此的结论。
IRAK4通过介导TLRs和IL-1R信号来激活NF-κ B。反过来,这会上调许多NF-κ B依赖性基因的转录,包括那些与炎症小体有关的基因[134]。近年来,已经在四家公司开展了IRAK4抑制剂的临床试验[135]。IRAK4抑制剂已经被证明可以通过干扰体外和体内的引发来抑制炎症小体的活性。在动物模型中,IRAK4抑制剂以展示出可以降低分泌的IL-1 β 水平,尽管它们也对其他促炎细胞因子(如IL-6)有影响,而这些细胞因子在炎症小体活化后不会分泌出来[136]。辉瑞公司开发的化合物PF-06650833,最近完成了一项关于类风湿性关节炎的IIb临床试验,并根据简化疾病活动指数从基线的变化来看是有效的[137](表3)。 然而,目前还没有一份详细介绍这些结果的完整公开数据。尚不知道IRAK4抑制剂对人的炎症小体基因表达具有何种程度的作用,以及这是否有助于临床效果。
脾酪氨酸激酶(Spleen tyrosine kinase,Syk)是一种非受体蛋白酪氨酸激酶,其传递造血细胞的B细胞抗原受体或Fc受体信号,并且与许多疾病密切关联,也与炎性小体活化有关。我们检查了Syk抑制对阻断炎症小体活化的影响,以观察Syk抑制剂的临床疗效或安全性信号是否与炎症小体的表型有关。Syk上的许多磷酸化位点的激活导致了与免疫和自身免疫疾病相关的一组信号网络的激活。因此,Syk是免疫细胞炎症反应的主要调节因子,抑制Syk活性可以广泛而全面地减少炎症[138]。最近的一项研究表明,Syk与IL-1 β的分泌有关,其机制与NLRP3炎症小体的分泌有关,NLRP3炎症小体在糖尿病肾病的炎症反应中起着传感器和调节因子的作用,导致pro-caspase-1裂解和IL-1 β 成熟。Syk的磷酸化可以提高pJNK的水平及其下游相互作用子NLRP3、caspase-1p20、ASC和mIL-1 β 在高糖诱导的HK2细胞和RGMCs中的表达[139]。此外,作者还发现,通过小分子BAY61-3606和Syk-siRNA抑制Syk能够阻断这种信号级联,减少IL-1 β的释放。其他文献报道也支持Syk信号在结核分枝杆菌感染和真菌感染期间与NLRP3功能和IL-1 β 的产生相关的关键作用[140,141]。近年来,12种口服小分子Syk抑制剂已进入炎性疾病适应症的临床研究,如哮喘、关节炎和过敏性疾病[142]。Syk信号与免疫细胞的激活有关,假设抑制Syk可以减轻自身免疫反应[140,143]。尽管早期有疗效信号,但许多II期和III期试验被撤回或终止而没有公开数据,我们缺乏公开披露的可说明安全性与治疗窗的临床数据。Syk抑制剂的发现和开发已被全面综述[138],详细介绍了Syk抑制的生物学、已公开的小分子抑制剂的概况、临床前模型和早期临床结果。另一项最新的研究涵盖了2010年至2013年间申请的Syk抑制剂及其在临床上的应用路径[142]。作者强调了专利公开的结构多样性和激酶选择性的差异。除了疗效外,临床上的失败也是未来靶向炎症小体的努力所感兴趣的,因为这些可能指出关键的安全性问题并予以避免,并证明与慢性炎症相关的疾病明显缺乏疗效。在进入临床的12种Syk抑制剂中,没有一种被推向市场治疗大量患者群体的疾病。
Fostamatinib(Tavalisse,R788)由Rigel发现,并由阿斯利康开发,是唯一上市的Syk抑制剂[144]。目前由Rigel在市场上销售用于治疗成人慢性免疫性血小板减少症(ITP)患者的血小板减少症[145]。Fostamatinib是第一种到达临床的Syk抑制剂,在457例活动性类风湿关节炎患者(尽管进行了甲氨蝶呤治疗)的II期试验中显示出了早期的希望。IIb期研究(TASki2)证明67%的每天两次服用Fostamatinib 100mg的患者在6个月时达到主要疗效终点,这显著高于安慰剂[146]。出现了副作用高血压,但认为用适当的药物治疗是可以控制的。这些令人兴奋的结果启动了一系列三期试验OSKIRA:口服SyK抑制剂用于类风湿性关节炎[147−150]。尽管100 mg每日两次(BID)组的ACR20反应率在24周内有统计学上的显著改善,并且在RA患者的OSKIRA三期项目中可耐受的安全性结果,阿斯利康没有继续进行监管备案,而是将Fostamatinib的权利归还给了Rigel。2019年,一篇旨在研究多剂量Fostamatinib安全性和有效性的系统性综述和荟萃分析发表了一个令人感兴趣的展望[151]。他们证实,在包括3680名患者在内的11项临床试验中,Fostamatinib是一种有效和安全的治疗药物。对于总体不良事件,发现在100mg BID的Fostamatinib和安慰剂之间存在显著差异。显著的不良事件包括胃肠道失调、肝胆失调、感染和传染以及肌肉骨骼和结缔组织失调。然而,与安慰剂组相比,在Fostamatinib组中没有发现严重感染和传染频数的显著增加。这一观察结果与使用Canakinumab患者严重感染的增加不一致,这可能是Syk抑制剂与炎症小体抑制的机制差异。由于只有中等的疗效和一些安全信号,人们担心Fostamatinib是否有能力在RA市场上与阿达木单抗和托法替尼竞争。
Fostamatinib缺乏激酶选择性是解释临床数据和我们努力将临床疗效或安全性与炎症小体抑制联系起来的关键。Fostamatinib(R788)是其活性代谢物R406的前药。已经有人对R406的体外药理学作用谱进行了全面的评估[152]。作者发现R406在治疗浓度下与多种激酶结合。从临床PK数据推断,他们设定了150nM的阈值,并识别出101个具有更强作用的R406靶标,其中只有一个不是激酶。 虽然Fostamatinib的主要机制可能是Syk抑制,但我们不能排除100种激酶抑制的脱靶或多药效应。
随着对Fostamatinib II期临床数据的兴奋,下一代Syk抑制剂的目标是更高的选择性、更强的药效和口服生物利用度。MK-8457,一种新颖的Syk和ZAP70(Zetachain相关蛋白激酶)抑制剂,在两个II期试验中由于严重感染率高而终止。由Biogen和Portola共同开发的Syk/JAK双重抑制剂Cerdulatinib(PRT062070,RVT-502,BIIB-057)报告了I期临床的健康志愿者PK数据和安全数据[153]。Cerdulatinib具有理想的PK特征,在每天口服一次后能够安全、强效,选择性地抑制人体内的Syk激酶功能。 随着一期临床试验的完成,系统性红斑狼疮(SLE)和类风湿性关节炎的二期临床试验已经计划好,但从未启动[154]。这些Syk抑制剂的终止表明存在不利的安全性信号,这可能会阻碍这些药物的进一步开发[151]。
总的来说,Syk抑制剂已经从早期的临床适应症(在2013年Fostamatinib失败之前,适应症主要针对哮喘、类风湿性关节炎和血小板减少症等具有大量患者群体的炎症适应症)转向了肿瘤适应症。包括Fostamatinib(R788)、Entospletinib(GS-9973)、Cerdulatinib(PRT062070)和TAK-659在内的Syk抑制剂正在临床试验中进行评估对B细胞恶性肿瘤、急性髓系白血病(AML)和慢性淋巴细胞白血病(CLL)的作用,并对血液学恶性肿瘤进行联合治疗方案[155](表3)。尽管有临床前证据表明Syk抑制与炎性小体激活的抑制有关,但很难断定任何疗效或安全性信号是否可归因于NLRP3炎症小体激活的减弱。
NLRP3与安全性
在许多方面,NLRP3似乎是理想的药物靶点,因为它在许多慢性炎症疾病中被异常激活。 这让人不禁要问,为什么NLRP3首先在进化上得以维持。没有已知的功能缺失(loss of function ,LOF)变体,并且其它物种的炎症小体传感器可相当大地变化。例如NLRP1,小鼠表达NLRP1a、NLRP1b和NLRP1c三个同源物,并且NLRP1b和NLRP1c的五个不同等位基因变体[156]。另一方面,NLRP3在不同的物种之间表现出相对保守性,并且对许多相同的刺激有反应。在蝙蝠中,NLRP3炎症小体的抑制与持续性的病毒库的存在有关[157]。在人类中,NLRP3对某些病原体的控制,尤其是RNA的病毒,可能是很重要的。
据报道,NLRP3炎症小体被甲型流感病毒感染(IAV)激活。激活是通过感知病毒RNA和流感病毒蛋白M2(一种离子通道)来实现的[158,159]。因此,NLRP3的抑制会影响宿主对IAV感染的反应[160]。当小鼠感染低浓度甲型流感并在感染后第二天MCC950治疗时,经MCC950治疗的小鼠在6天后死亡,而空白药治疗的小鼠或用MCC950治疗但未感染的小鼠直到第8天、以及本实验研究的最后一天没有任何死亡率。随着病毒接种浓度的提高,MCC950治疗组和空白治疗对照组的小鼠之间的差异变小。WT小鼠在第5天死亡,而经MCC950治疗的小鼠在第4天死亡。另一方面,用高剂量甲型流感感染然后用MCC950治疗的动物表现出更好的结果。这可能是由于预防细胞因子风暴和随后的NLRP3反应过度活跃对肺组织的损伤。这些数据表明NLRP3活动对感染初始反应产生的重要性。作者认为,一旦感染已经发展,使用NLRP3抑制剂的情况更有可能是临床场景。使用NLRP3抑制剂来减弱对已经发生感染的先天免疫应答,这可能是正确的。 然而,在慢性疾病中,如帕金森氏症或NASH,患者可能服用NLRP3抑制剂多年,目前还不清楚这对流感感染会有什么影响。一旦观察到感染,就有可能需要终止小分子药物治疗。例如,MCC950的半衰期只有三个半小时,尽管这种方法的临床可行性以及它对疾病进程的影响还没有研究。
为了支持这些数据,对NLRP3 敲除(KO)小鼠的研究也显示出对IAV感染的反应较小,死亡率增加[159,161]。在IAV感染后,NLRP3 KO动物的中性粒细胞、单核细胞和树突状细胞的补充减少,上皮坏死增加,坏死碎片导致肺阻塞。KO和WT之间的病毒滴度在感染后第3天、感染后第7天是相似的,WT动物比NLRP3 KO小鼠具有更低的病毒滴度。Caspase-1 KO动物显示相似的作用,而NLRC4 KO动物与WT动物无显著差异。与NLRP3对IAV感染反应的重要作用相一致,NLRP3 GOF R258W突变动物对IAV感染有抵抗力[162]。这些动物在IAV感染后存活率提高,IL-1R敲除后丧失对IAV感染的应答。同样,老年小鼠在IAV感染后先天免疫反应降低,存活率较差[163]。用尼吉霉素(Gigericin)刺激可增加其炎症小体活性,并改善IAV感染后的结果。总之,这些数据表明在IAV感染时功能性NLRP3激活对于建立成功的免疫应答是重要的。
NLRP3炎症小体也被证明对肺炎链球菌(S.P.)感染的反应很重要[164,165]。在老年小鼠中,NLRP3敲除导致S.P.感染后的死亡率更高[166]。一般来说,老年小鼠的炎症小体激活受损,对肺炎链球菌感染的应答更差。牛磺熊去氧胆酸可挽救这种情况,它是一种ER分子伴侣,能上调NLRP3、ASC、caspase-1和IL-1 β 的分泌。年龄大的小鼠对IAV感染后的继发S.P.感染的反应也很差[167]。一般来说,先天免疫功能似乎随着年龄的增长而下降。虽然基础活性似乎增加,但诱导型活性似乎在减少[168]。
NLRP3也在防御真菌感染中起作用。白色念珠菌已清楚地证明了这一点。真菌肽canderalysin通过钾外流激活小鼠和人巨噬细胞中的NLRP3炎症小体[169]。NLRP3 KO增加了小鼠白色念珠菌感染后的菌落形成单位(CFU)数量[141,170,171]。在小鼠感染模型中,在没有NLRP3或其他炎症小体成分(如IL-1R1、ASC或Caspase-1)的情况下[141,170],存活率降低。然而,这些研究没有使用药物抑制剂,这限制了将这些结果外推到临床场景的能力。
研究还表明NLRP3炎性染色体在适应性免疫中的作用。树突状细胞和T细胞也有炎症小体激活。Ghiringhelli等人[172]指出,在癌细胞死亡时,树突状细胞中的NLRP3炎性细胞团被激活。反过来,这又触发了IL-1 β 的分泌、CD8+T细胞的引发和IFN-γ的分泌。与WT相比,在奥沙利铂治疗后,敲除炎症小体组分NLRP3、P2RX7或Caspase-1会导致肿瘤的体积增加。同样,在CD4+细胞中,IFN-γ 的分泌以及TH1的反应需要NLRP3的功能[173]。 MCC950治疗可阻止TH1细胞分泌IFN-γ,Caspase-1抑制剂也是如此。此外,CAPS患者CD4+T细胞分泌IL-1 β 和IFN-γ增加。NLRP3抑制剂可降低IL-1 β 和IFN-γ 的分泌。 NLRP3 KO小鼠CD4+T细胞的CD3/CD28激活在体外导致IFN-γ 分泌量仅达到WT小鼠CD4+T细胞在体外分泌量的25%。在对淋巴细胞性脑膜炎病毒感染的应答中,CD4+T细胞缺乏NLRP3、IL-1-R1或IL-1 α/IL-1 β,表现出体内产生IFN-γ+病毒特异性细胞的能力受损。这些数据指出了这种途径作为对病原体感染的先天和适应性免疫反应的一部分的重要性。这有助于解决NLRP3炎症小体的生物学用途是什么的问题。这也凸显了在这一令人兴奋的靶标进行新药测试时谨慎行事的必要性。
NLRP1炎症小体
除了NLRP3之外,还有许多其他炎症小体传感器,其中许多与疾病紧密关联。一般来说,这些都超出了本综述的范围;然而,NLRP1炎症小体与NLRP3的一些无菌炎症紧密相关,因此值得特别提及。到目前为止,尚无已知的NLRP1小分子工具化合物。在临床前领域,IFM公开了一种调节NLRP1、NLRP3和NLRP1/3双调节化合物。然而,没有显示NLRP1抑制的数据[174]。有报道称,肽KSHV Orf63显示出抑制NLRP1的能力。人类NLRP1的病毒同源物KSHV Orf63阻断NLRP1依赖性先天性免疫应答,包括Caspase-1激活和白细胞介素IL-1 β 和IL-18的加工。 KSHV Orf63不是NLRP1选择性的,并且还显示与NLRP3和NOD2相互作用[175]。GSK的文章提出了识别NLRP1抑制剂的策略。作者对NLRP1 ATP-竞争性抑制剂进行了高通量筛选,并在荧光偏振分析法发现了苗头化合物,但没有在这些苗头化合物中发现在细胞水平有活性的化合物[176]。与围绕NLRP3的大量专利和公开文献相比,研究NLRP1抑制领域的能力较低,并且缺乏一种小分子工具化合物来探索其生物学。NLRP3之外的炎症小体小分子工具化合物的发现有可能为炎症小体的生物学开辟一个新的前沿学科。
结论
NLRP3炎症小体是一个很有前途的药物靶点。它与众多的适应症紧密相关说明了这一通路具有显而易见的中心地位。在接下来的几年里,随着最先进的临床研究开始给出疗效数据,科学家、医生和患者都将拭目以待,看看这些期望是如何得到证实的。抑制人类NLRP3炎症小体的明确保护功能还没有被证实。基于NLRP3的保守性,由于在人类中没有NLRP3功能缺失(LOF)变体的描述,一些化合物感染严重副作用、临床前物种的某些类型感染的易感性、NLRP3抑制可能会产生的副作用等等报道可能阻碍炎症小体的药物开发。同时,基于抑制炎症小体激活或抑制炎症小体激活下游产物的化合物药效数据,有望通过避开直接抑制NLRP3本身的新机制或通路来抑制炎性小体。最终,这将是未来几年生物学和药物发现的一个令人兴奋的领域。
行业新闻
- 2021.04.12 NLRP3炎症小体候选药物BMS-986299结构首次公开

Leigh Krietsch Boerner(2021.04.12). Virtual meeting delivers first time drug structures. Retrieved from https://cen.acs.org/acs-news/acs-meeting-news/Virtual-meeting-delivers-first-time-drug-structures/99/web/2021/04


