
摘要:帕克替尼(Pacritinib)是一种可口服的、靶向JAK2的大环类抑制剂,在治疗骨髓纤维化方面已显示出疗效。由于合成大环类化合物存在合成挑战,我们应用了静电互补性分析、3D-Field QSAR以及自由能微扰(FEP)等方法,对一组已知的大环帕克替尼衍生物进行了综合评估,旨在建立优先级排序方法用来选择新的大环类化合物以进行合成。研究还表明,理解配体的3D构象和柔性至关重要,这些因素对预测的准确性具有显著影响。
原文:Braz, N.F., Slater, M.J. and Lang, S. (2025) “Application of Free Energy Perturbation (FEP) Methodology for Predicting the Binding Affinity of Macrocyclic JAK2 Inhibitor Analogues of Pacritinib,” ACS Medicinal Chemistry Letters. Available at: https://doi.org/10.1021/acsmedchemlett.5c00105.
编译:肖高铿/2025-06-02
大环化合物可以提供半刚性、预组织好的骨架,通过“系链”(tether)将两个远离的基团连接在一起,使其彼此靠近,从而限制了小分子配体的全局构象柔性1。与等效的开环类似物相比,大环化合物在结合亲和力和选择性方面带来显著改进,尽管它们可能具有超出类药五规则(bRo5)的性质,但其中许多仍具有足够的细胞渗透性,能够实现口服生物利用度2。大环结构广泛存在于许多具有生物活性的天然产物中,其中部分已被批准为药物。近年来,合成设计的大环化合物也逐渐成为具有治疗潜力的重要化合物类别3。
尽管大环化合物在药物发现中提供了有吸引力的设计前景,但与其开发相关的合成挑战限制了它们的应用推广4。虽然一些金属催化的反应,如关环复分解反应(RCM)5、Suzuki-Miyaura 反应6、Heck 反应7以及炔烃-叠氮点击反应8等,提高了大环化合物的可合成性,但由于这些反应通常需要在低浓度下进行,以尽量减少聚合副反应的发生,因此其应用并不简单9。此外,由于大环化步骤通常需要在合成路线的后期进行,因此结构多样性的引入往往需要在其早期阶段就整合到分子骨架中10。为了平衡这种在合成资源上更高投入所带来的风险,在设计规划阶段就需要对项目构思进行更严格的审查,以确保这些努力不会徒劳无功11。
由于这些挑战,我们对应用计算机辅助药物设计(CADD)工具来评估具有生物活性的大环类化合物感到非常兴奋。JAK2是一个重要的治疗靶点,其活性与多种血液系统疾病相关,例如骨髓增殖性肿瘤(MPNs)12,目前已有4个JAK2抑制剂获得FDA批准(见图1):鲁索替尼(ruxolitinib,1)13、菲卓替尼(fedratinib,2)14、莫美洛替尼(momelotinib,3)15和帕克替尼(pacritinib,4)16。其中,帕克替尼(4)的大环结构特别引起我们的兴趣。结合近期解析的JAK2-帕克替尼复合物结构(PDB 8BPV)17,我们认为这是一个开展本研究的理想对象。
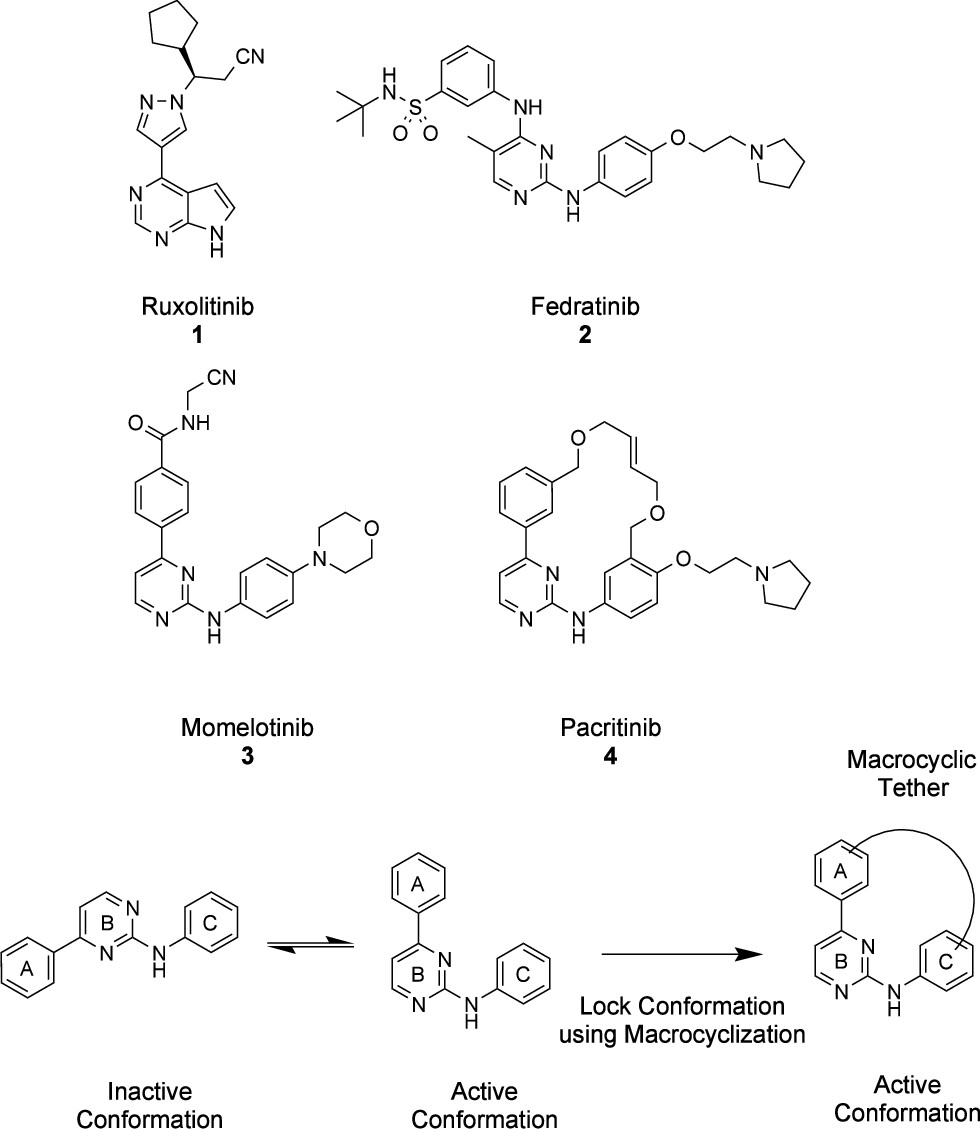
图1. JAK2抑制剂鲁索替尼(1)、菲卓替尼(2)、莫美洛替尼(3)和帕克替尼(4),以及化合物3和4在活性与非活性构象中的ABC环骨架结构。
莫美洛替尼(3)和帕克替尼(4)共有一个由ABC环系组成的结构,其中芳香A环直接连接到嘧啶B环的4位,B环的2位通过NH基团连接至芳香C环。这两个化合物之间唯一的结构差异在于,帕克替尼(4)引入了一个将A环和C环连接起来的连接臂,而莫美洛替尼(3)没有该连接臂。这个连接臂将帕克替尼(4)锁定在其适合与JAK2结合的活性构象,防止其转变为非活性构象(如图1所示)。该非活性构象可能导致与其它无关靶点的结合,从而引发选择性方面的问题。
在JAK2–帕克替尼复合物结构中,帕克替尼(4)(见图2)与铰链区和柔性P-loop的Leu932、Leu855发生相互作用,并通过水分子介导的方式与Ser936和Asp939形成氢键。通过3D-RISM方法分析发现,介导这些相互作用的水分子是稳定的,其自由能(ΔG)分别为−4.2、−11.8 和 −1.6 kcal/mol。帕克替尼(4)还稳定了折叠状态的P-loop结构,在该构象中,Phe860的侧链在与抑制剂结合后深埋入到ATP结合口袋。这种折叠的P-loop构象,加上P-loop中的Asn859与DFG中的Asp994之间缺乏分子内氢键,被确定为JAK2选择性的结构决定因素。这一特征解释了帕克替尼(4)所表现出的中等程度的JAK2选择性,对JAK2的选择性比其他JAK家族成员高\(≥6\)倍17。
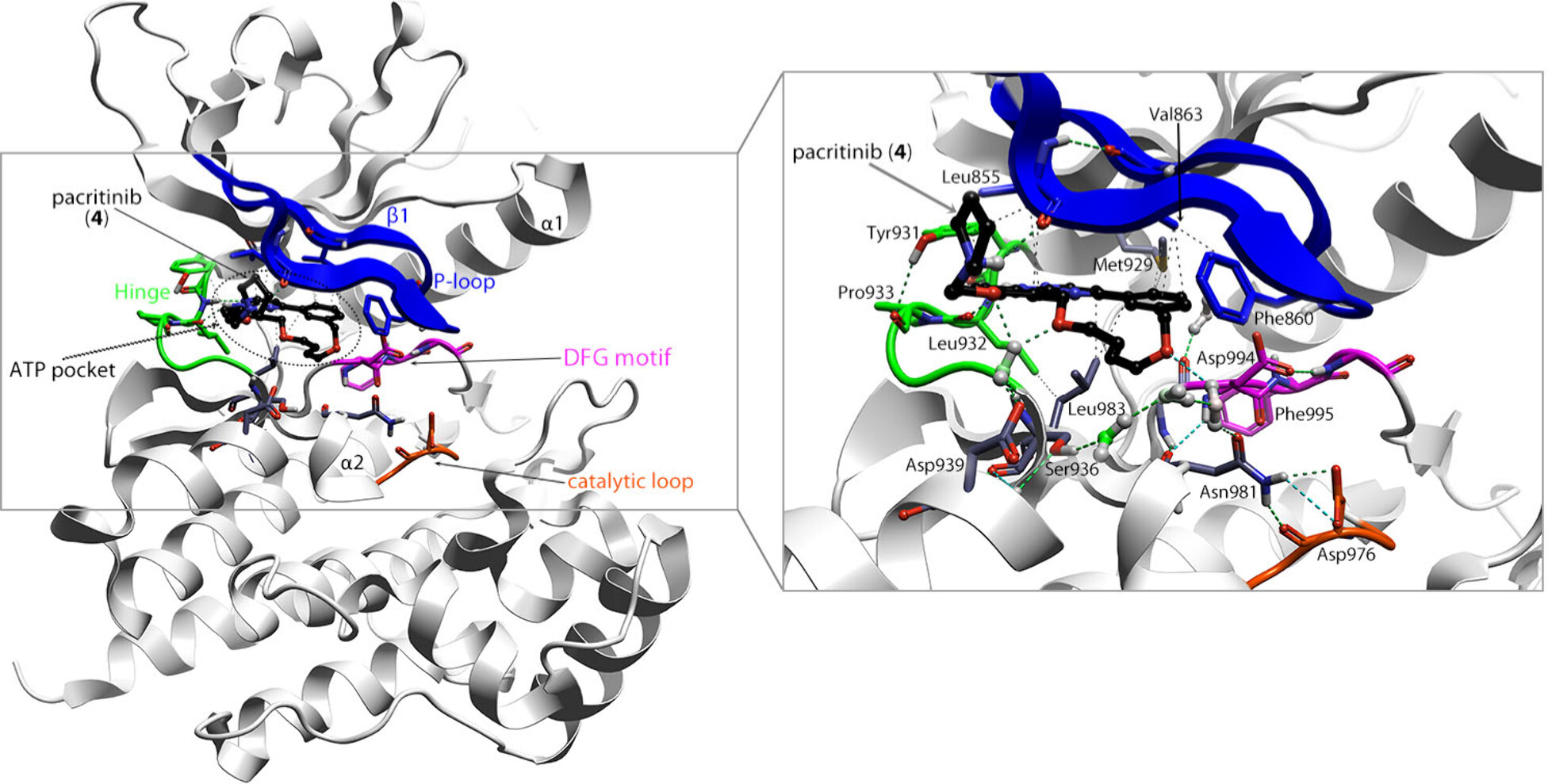
图2. 帕克替尼(4)在JAK2的ATP结合位点中的结合模式。
计算机辅助药物设计(CADD)方法可以显著减少探索化合物系列构效关系(SAR)所需的时间和资源18。准确预测蛋白质–配体结合亲和力已成为该技术的一项重要应用,提高了获得新候选药物的成功概率。先前的研究报道了一系列帕克替尼(4)的大环衍生物19。由于这些化合物具有高度的结构相似性,预计它们会采用与最近解析的帕克替尼(4)相似的结合模式。本文中,我们讨论了帕克替尼(4)以及一组大环类似物(图3)的结合分析。
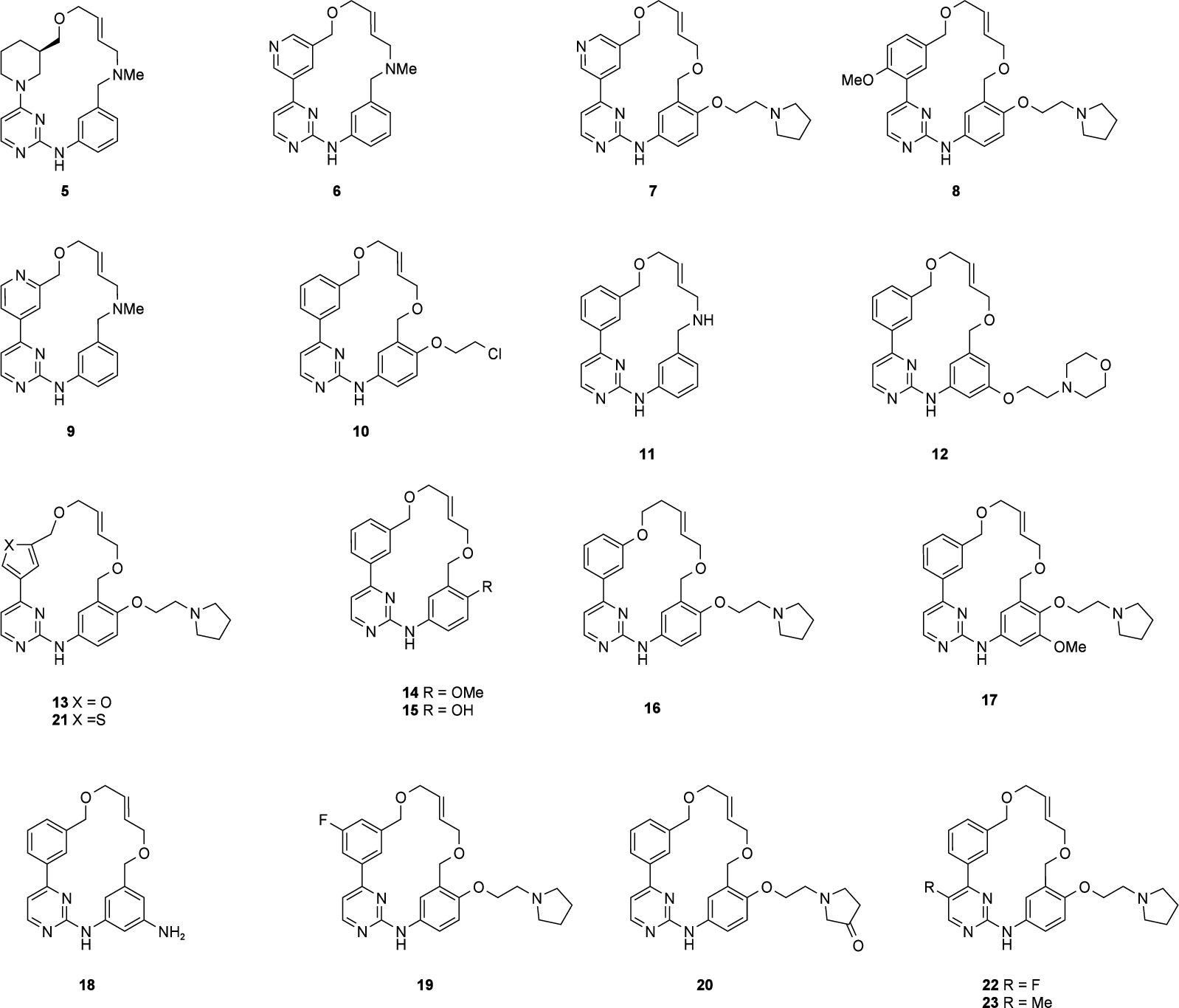
图3. 帕克替尼(4)的大环类似物。
这些类似物探索了将芳香A环替换为多种杂环结构(化合物5、6、7、9、13和21),以及在邻位(化合物 8)和间位(化合物 19)引入取代基的影响。同时,还研究了在芳香C环的间位上增加取代基(化合物12、17和18),以及在对位上改变水溶性链段(化合物10、14、15和20),并在嘧啶B环的5位引入取代基(化合物22和23)。最后,还考察了对大环连接臂进行修饰的类似物(化合物5、6、9、11和16)。
作为初步分析,我们将实验测得的pIC50值与计算得到的静电互补性(EC)20值进行了比较,所用工具为Flare V10中的实现,用于评估配体与蛋白质之间的匹配程度。该分析涵盖了所有研究中的配体,并基于X-衍射结构(PDB 8BPV)中帕克替尼(4)的生物活性构象(图2),采用以配体为中心的分子叠合方式进行。令人欣慰的是,该方法能够有效识别哪些化合物可能不具有活性:那些EC打分低于0.29的化合物,其pIC50值也都低于7.0。这一标准成功预测了75%的实验pIC50 \(<\) 7.0的化合物。该指标的适用性也通过其较高的Kendall’s Tau和Pearson相关系数得到了验证,分别为0.46和0.75。
化合物7和8与帕克替尼相比,仅在A环的西边部分存在结构差异,它们均表现出良好的EC打分(分别为0.36和0.32),但其pIC50值较为一般(均为6.5)。在化合物7中引入吡啶氮原子使LogP降低了0.6。然而,与帕克替尼(4)相比,这一改变导致pIC50下降了1.1,意味着亲脂配体效率(LLE)总体下降了0.5。EC能够将打分较低的配体合成优先级降低,从而提供一种非常快速的方法,可显著减少原本合成复杂化合物所需的工作量。
在此之后,我们希望应用能够生成具有预测性的pIC50值的方法,以对化合物进行排序。采用与生成EC打分相同的分子叠合方法,并使用Flare V10中实现的3D Field-QSAR模型21,我们构建了一个包含69个帕克替尼类似物的QSAR模型,这些配体的活性分布均匀(pIC50范围为5.0至8.2)。我们将这些化合物划分为训练集(49个配体)和测试集(20个配体,见表1)。有关该QSAR模型的完整细节,请参见支持信息。
表1. Pacritinib及其大环衍生物的pIC50实验观察值与预测值
| Comp. | Calc. LogP | Exp. pIC50 | LLE | EC | 3D-QSARpred. pIC50 | FEP (Open FF)pred. pIC50 |
|---|---|---|---|---|---|---|
| 4 | 4.8 | 7.6 | 2.8 | 0.36 | 7.6 (−) | 7.5 (−0.1) |
| 5 | 3.3 | 5.0 | 1.7 | 0.19 | 6 (+1.0) | 5.1 (+0.1) |
| 6 | 3.7 | 5.0 | 1.3 | 0.27 | 5.1 (+0.1) | 5.4 (+0.4) |
| 7 | 4.2 | 6.5 | 2.3 | 0.36 | 6.6 (+0.1) | 6.9 (+0.4) |
| 8 | 4.8 | 6.5 | 1.7 | 0.32 | 7.4 (+0.9) | 6.5 (−) |
| 9 | 3.7 | 6.6 | 2.9 | 0.26 | 5.9 (−0.7) | 5.7 (−0.9) |
| 10 | 5.0 | 6.7 | 1.7 | 0.27 | 6.7 (−) | 7.3 (+0.6) |
| 11 | 4.2 | 6.8 | 2.6 | 0.27 | 6.0 (−0.8) | 6.6 (−0.2) |
| 12 | 4.1 | 7.0 | 2.9 | 0.29 | 7.3 (+0.3) | 7.9 (+0.9) |
| 13 | 3.8 | 7.2 | 3.4 | 0.37 | 7.2 (−) | 7.5 (+0.3) |
| 14 | 4.4 | 7.2 | 2.8 | 0.34 | 7.5 (+0.3) | 7.1 (−0.1) |
| 15 | 4.0 | 7.3 | 3.3 | 0.34 | 7.4 (+0.1) | 6.8 (−0.5) |
| 16 | 5.1 | 7.3 | 2.2 | 0.36 | 7.4 (+0.1) | 7.4 (+0.1) |
| 17 | 4.8 | 7.4 | 2.6 | 0.31 | 7.5 (+0.1) | 7.5 (+0.1) |
| 18 | 3.9 | 7.5 | 3.6 | 0.31 | 7.1 (−0.4) | 7.3 (−0.2) |
| 19 | 5.2 | 7.6 | 2.4 | 0.37 | 7.3 (−0.3) | 7.7 (+0.1) |
| 20 | 4.0 | 7.7 | 3.7 | 0.33 | 7.5 (−0.2) | 7.5 (−0.2) |
| 21 | 4.9 | 7.8 | 2.9 | 0.37 | 7.2 (−0.6) | 8.1 (+0.3) |
| 22 | 5.2 | 7.8 | 2.6 | 0.35 | 6.9 (−0.9) | 7.5 (−0.3) |
| 23 | 5.1 | 8.2 | 3.1 | 0.35 | 7.5 (−0.7) | 7.1 (−1.1) |
括号里的“deviation”数值指的是计算得到的pIC50值与实验测得的pIC50值之间的差异。
该模型性能表现良好,训练集的r2 = 0.92,训练集的交叉验证q2 = 0.47,测试集的r2 = 0.52。测试集获得的Kendal’s Tau 相关系数为 0.45。在本研究的许多化合物中,该QSAR模型预测的活性与真实实验值之间的误差在0.5个对数单位以内,但在活性范围两端的一些化合物中,预测值与实验值之间的差异可能高达1个对数单位。
为了获得更深入的认识,我们决定在配体分析中使用Flare V10中实现的自由能微扰(FEP)方法22。据我们所知,目前将FEP应用于大环化合物预测的案例还很少见23。令人欣慰的是,在使用Open Force Field进行计算时,我们观察到在大多数情况下,使用FEP后预测的pIC50值与实验值之间的差异在0.5个对数单位以内,Kendall’s Tau和Pearson相关系数分别为0.6和0.86。进一步分析那些与实验结果偏差较大的配体发现,化合物9涉及电荷变化(O变为NMe),这在FEP计算中更具挑战性。另一个与实验活性偏差较大的配体是化合物12,它涉及将四氢吡咯环替换为吗啉环。由于这一变化发生在配体的增溶区域,可能意味着该部分与蛋白的相互作用较弱,并且在整个分子动力学(MD)模拟过程中存在波动。因此,这种预测不如那些直接影响配体-蛋白结合的修饰那样准确,也就不足为奇了。
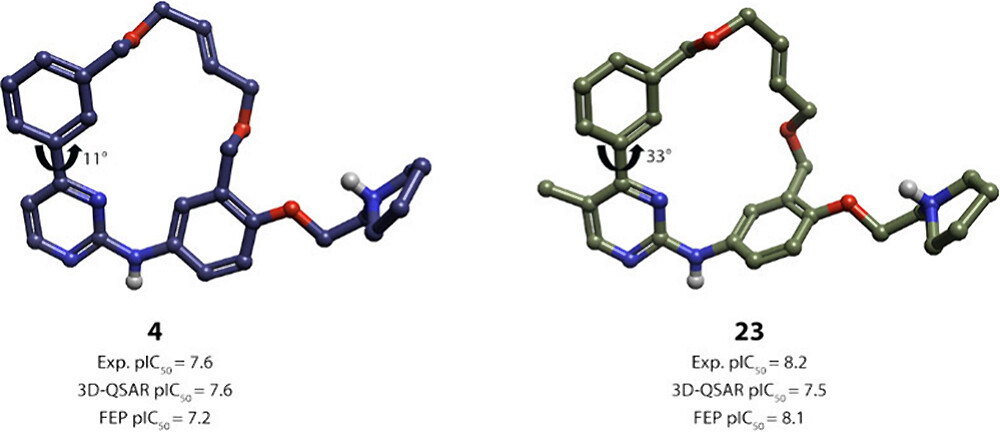
与实验测量的pIC50相比,化合物23的活性通过FEP方法预测时显著偏低,计算得到的pIC50值比实验值低1.1。由于该化合物是我们研究中活性最高的配体,我们决心找出这一较大偏差的原因。我们推测,在嘧啶B环的5位添加一个甲基会导致AB双芳基键的旋转,从而改变化合物23相对于帕克替尼(4)晶体结构结合模式下的优势轴向取向25。
因此,我们采用了量子力学(QM)方法为化合物23生成了其他可能的构象,并从中选出了4种结构多样且能量较低的构象用于进一步分析(图4)26。最初的构象23A是通过与晶体结构进行严格叠合生成的,其A环和B环之间的二面角为18°。而在新生成的低能构象中,由于甲基的存在,该角度有所增加,分别为23B:26°,23C:24°,23D:33°。
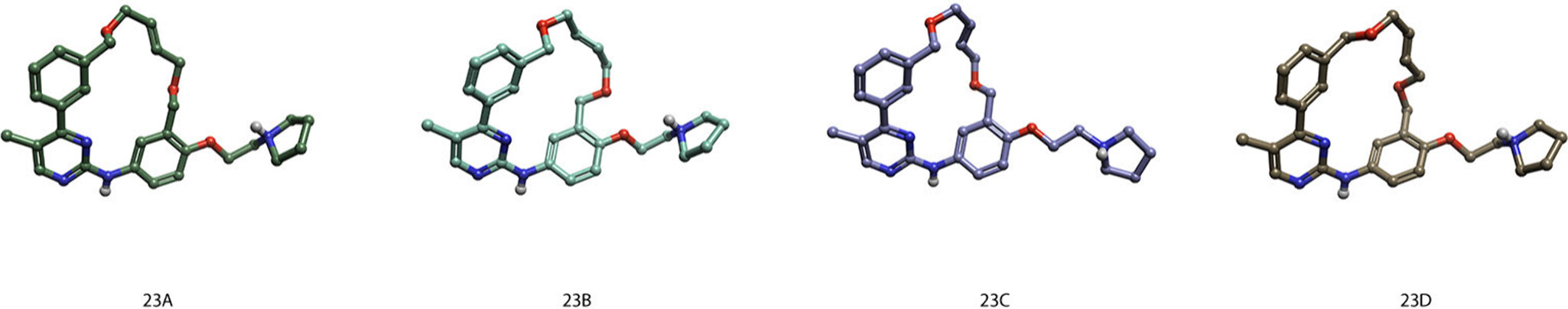
图4. 化合物23的不同低能构象的3D示意图。
尽管大环体系可以用来限制整体的构象变化,但AB连芳基环系的构象改变会引发大环内部其他二面角的一系列连锁变化。例如,位于A环和C环区域的苄基醚键能够自由旋转,以适应AB环系受限的运动。该柔性使得在建模特定构象时需要格外谨慎27。
采用环状图(cyclic map)对化合物23的合种构象、帕克替尼(4)以及化合物22 进行了FEP基准研究,以验证化合物23的优势构象(见表2)。虽然构象23-D的计算pIC50值比实验值低0.3,但它确实预测该化合物比其他测试的化合物更具活性。不仅FEP计算结果显示23-D是最优构象,而且其构象能也是最低的,比23-A的构象能低8.1 kcal/mol。
表2. 用FEP分析验证化合物23的构象
| Comp. | Exp. pIC50 | Pred. pIC50 with 23A | Pred. pIC50 with 23B | Pred. pIC50 with 23C | Pred. pIC50 with 23D |
|---|---|---|---|---|---|
| 4 | 7.6 | 7.9 (+0.3) | 7.8 (+0.2) | 7.8 (+0.2) | 7.8 (+0.2) |
| 22 | 7.8 | 8.0 (+0.2) | 7.9 (+0.1) | 7.9 (+0.1) | 7.8 (−) |
| 23 | 8.2 | 7.7 (−0.5) | 7.8 (−0.4) | 7.8 (−0.4) | 7.9 (−0.3) |
括号里的“deviation”数值指的是计算得到的pIC50值与实验测得的pIC50值之间的差异。
尽管我们对化合物23的D构象较为有信心,但为了进一步提高pIC50预测值与实验值之间的相关性,仍需要进行更多的优化。为此,我们对FEP的力场进行了变化(见表3),或在平衡过程中引入了“大正则系综非平衡候选蒙特卡洛”(Grand Canonical Nonequilibrium Candidate Monte Carlo,简称GCNCMC)方法28(见表3)。令人欣慰的是,对于化合物4、22和23来说,当配体使用GAFF力场29时得到了与实验值高度一致的结果,其表现优于Open FF(无论是否加入了自定义参数)30,也优于通过GCNCMC改善溶剂化环境的方法。
表3. 使用不同方案优化FEP预测结果
| Comp. | Exp. pIC50 | Pred. pIC50Open FF | Pred. pIC50GAFF | Pred. pIC50Open FF(custom parameters) | Pred. pIC50Open FF(GCNCMC) |
|---|---|---|---|---|---|
| 4 | 7.6 | 7.8 (+0.2) | 7.6 (−) | 7.8 (+0.2) | 7.8 (+0.2) |
| 22 | 7.8 | 7.8 (−) | 7.8 (−) | 7.8 (−) | 7.9 (+0.1) |
| 23 | 8.2 | 7.9 (−0.3) | 8.1 (−0.1) | 7.8 (−0.4) | 7.8 (−0.4) |
括号里的“deviation”数值指的是计算得到的pIC50值与实验测得的pIC50值之间的差异。
在完成这一优化过程后,我们将最优力场(配体上使用GAFF)应用于全部20个化合物的FEP基准研究中(见表4)。Kendall’s Tau和Pearson相关系数分别为0.55和0.84。在大多数情况下,如今pIC50的预测值与实验测得值之间的偏差控制在了0.5以内,其中GAFF对活性更高的化合物表现出更准确的预测能力。进一步分析那些偏差较大的化合物发现,像化合物9和11这类发生电荷变化的分子尤其具有挑战性。与化合物23类似,化合物8也含有一个会引发构象变化的基团(-OMe)25。
表4. 化合物4-23的FEP预测值
| Compound | Exp. pIC50 | Pred. pIC50FEP GAFF |
|---|---|---|
| 4 | 7.6 | 7.2 (−0.4) |
| 5 | 5 | 5.4 (+0.4) |
| 6 | 5 | 4.9 (−0.1) |
| 7 | 6.5 | 7.3 (+0.8) |
| 8 | 6.5 | 7.1 (+0.6) |
| 9 | 6.6 | 5.9 (−0.7) |
| 10 | 6.7 | 7.1 (+0.4) |
| 11 | 6.8 | 6.2 (−0.6) |
| 12 | 7.0 | 8.1 (+1.1) |
| 13 | 7.2 | 7.3 (+0.1) |
| 14 | 7.2 | 6.8 (−0.4) |
| 15 | 7.3 | 6.8 (−0.5) |
| 16 | 7.3 | 7.4 (+0.1) |
| 17 | 7.4 | 7.4 (−) |
| 18 | 7.5 | 7.4 (−0.1) |
| 19 | 7.6 | 7.4 (+0.2) |
| 20 | 7.7 | 7.3 (−0.4) |
| 21 | 7.8 | 7.8 (−) |
| 22 | 7.8 | 7.4 (−0.4) |
| 23 | 8.2 | 8.1 (−0.1) |
括号里的“deviation”数值指的是计算得到的pIC50值与实验测得的pIC50值之间的差异。
在嘧啶B环的5位引入甲基不仅改变了化合物23相对于帕克替尼(4)所采用的最低能量构象。通过10纳秒的分子动力学(MD)模拟研究比较连接A环和B环的键周围的旋转范围,结果显示化合物23的二面角最小为27°,最大为95°。而帕克替尼(4)不仅具有更大的旋转范围(−78° 至 37°),还能够采用正的和负的二面角构象。
尽管甲基的引入降低了A环与B环之间连接键的旋转柔性,但这并未有效减少大环连接臂的柔性。在10纳秒的模拟过程中,该连接臂呈现出多种不同的构象,尤其是在与C环相连的苯甲醚基团的取向方面尤为明显。毫不令人意外的是,最大的构象柔性出现在将水溶性四氢吡咯烷基团连接到C环的链段上。由于这一区域暴露在溶剂中,导致其结合亲和力的预测准确性较低,化合物12的情况就是其中一个例子。
总之,我们已经证明,计算机辅助药物设计(CADD)可用于对具有合成挑战性的配体进行优先级排序,其中EC、3D Field-QSAR和FEP方法均提供了有价值的见解。这些方法不仅对蛋白质体系的准备敏感,更重要的是也对所研究的配体结构非常敏感。在使用配体叠合方法时需要格外谨慎,因为并非所有类似物都会采用单一或相似的构象。通过详细的分析,可以提出优选的构象,从而实现对结合亲和力的准确预测。FEP方法的动态特性意味着它能对蛋白质和配体进行更充分的采样,而EC和3D Field-QSAR则是基于单一构象进行分析。尽管像EC和3D Field-QSAR这类计算成本较低的方法有助于配体合成的优先级排序,但FEP在预测结合亲和力方面具有更高的准确性。目前,我们团队正在进一步将这一综合平台应用于其他靶标体系,特别是那些需要复杂或耗时较长合成策略的目标化合物。
安全声明:本文不涉及任何具有危险材料的实验内容。研究过程中未遇到任何意外或异常的安全风险。
数据可用性
ZIP文件中的FLR文件可以使用Cresset公司的Flare软件打开,且可获取免费的可视化许可:https://www.cresset-group.com/software/licensing-flare
支持信息
支持信息可这里获取:https://pubs.acs.org/doi/10.1021/acsmedchemlett.5c00105
- 用于构建模型的支持性数据的文件夹:ml5c00105_si_001.zip
- 额外的实验细节:计算方法以及详细的补充结果,包括分子动力学(MD)模拟、结合口袋检测、水分子分析、3D-QSAR模型、FEP计算,以及实验值与计算机模拟值之间的相关性图谱。ml5c00105_si_002.pdf
文献
略
联系我们
如果您希望在您的项目中试用Flare的QM、MD、水分析、FEP、EC与3D-Field QSAR,请立即申请免费评估版的Flare。
- 电邮:info@molcalx.com
- 电话:020-38261356


