
计算方法用于配体-蛋白质相互作用排序和结合亲和力预测——哪种方法适合您?
摘要:本文系统综述了计算化学在药物发现中用于评估配体–蛋白相互作用和优化配体的主要方法。首先介绍了分子对接和静电互补性(EC)等快速、低成本的初筛技术,适用于项目早期配体优先级排序。随后讨论了基于实验构效关系(SAR)的3D-QSAR与Activity Atlas等定量/定性模型,强调其对数据质量和配体多样性依赖性强。文章重点阐述了自由能微扰(FEP)作为结合自由能预测的金标准,包括相对FEP(高精度,适用于同系列优化)与绝对FEP(适用于新骨架识别),并指出其计算成本高但预测能力优越。最后,文章提出应根据项目阶段、可用数据、目标及计算资源,合理选择或组合不同策略,并展望了AI/ML与主动学习在提升计算效率中的潜力。合理运用这些方法可显著降低实验成本、加速决策进程,推动药物研发向“先计算、后实验”范式演进。
原文:Lang, S. et al. (2025) “In Silico Methods for Ranking Ligand–Protein Interactions and Predicting Binding Affinities: Which Method is Right for You?,” Journal of Medicinal Chemistry, 68(19), pp. 19795–19799. Available at: https://doi.org/10.1021/acs.jmedchem.5c02582.
编译:肖高铿
衡量配体与蛋白结合的能力是所有药物发现项目的核心支柱,既包括最大化对主要靶点的亲和力,也包括通过最小化与非靶标蛋白的结合来提高选择性1。配体优化涉及多轮“设计−合成−测试−分析”(design−make−test−analysis, DMTA)循环,通常需要合成并进行多种生物测定以评估数百甚至数千个化合物,才能将候选药物推进至临床阶段。
配体–蛋白相互作用分析方法
评估配体与蛋白结合最简单且广为人知的方法之一是分子对接1 。该方法主要用于预测配体在三维空间中如何与蛋白结合。其计算速度快、成本低。对接打分以吉布斯自由能(ΔG)形式报告,负值越大表示配体与指定蛋白的结合能越强。对接算法通常为每个配体生成多个结合模式(pose),但打分最高的结合模式未必是正确的那个。因此,不能仅凭对接打分判断结合模式的合理性。
若已有已知活性的化合物,一种有效的对接验证方案是在所研究的蛋白中对这些配体生成带打分值的结合模式。这使得对接打分可与真实实验值进行比较,从而判断这些对接打分和/或所提出结合模式的有效性。通过对已知配体进行此类评估,可优化对接流程,并在合成前提升该打分方法用于新配体优先级排序的信心。
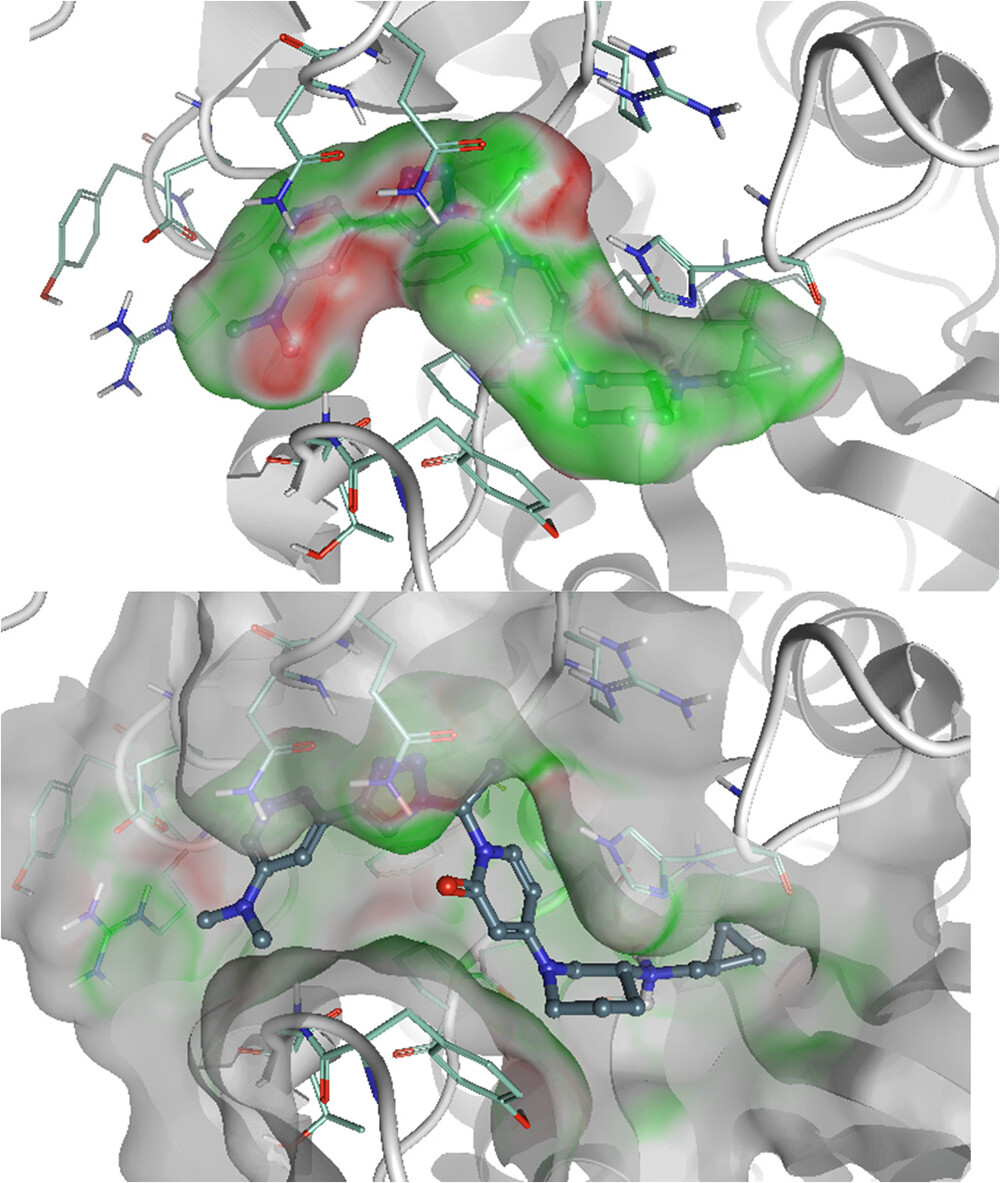
图1. 静电互补性(EC)表面添加到METTL3抑制剂(P6562)上,突出显示有利(绿色)和不利(红色)区域。
对接打分具有累加性,即与蛋白形成更多相互作用的大分子通常比小分子打分更高。因此,可采用对接配体效率(Ligand Efficiency, LE)等指标(如实验LE),以实现不同尺寸分子间的比较。静电互补性(Electrostatic Complementarity, EC)是一种低成本方法,利用扩展电子分布(eXtended Electron Distribution, XED)7 力场生成详细的分子表面映射,并提供定量分数,描述配体在蛋白口袋中的契合程度。与对接不同,EC并非基于配体与蛋白相互作用的数量,而是更强调相互作用的质量。在EC中,打分为1表示完美的静电互补,打分为−1表示完全的静电冲突。与对接类似,该分数基于单一配体和蛋白构象生成,因此除用于对接配体的排序外,也是一种快速且低成本的方法,可在系统分析初期用于评估所提出的结合模式。
EC打分不仅对蛋白和配体的静电性质敏感,也对溶剂敏感。因此,为获得准确打分,需了解结合口袋内的水分子网络。
尽管EC打分是在整个配体背景下计算的,但仍可生成表面图,显示局部区域的有利(绿色)或不利(红色)情况。出于可视化目的,该表面可叠加在配体或蛋白上,如METTL3抑制剂所示(图1)8。该图有助于选择配体的特定区域进行优化,以提高其与蛋白的互补性。
MM/GBSA(Molecular Mechanics/Generalized Born Surface Area)方法用于计算配体与蛋白的结合能,顾名思义,其基于分子力学,相比对接能更准确地描述结合能。与分子对接类似,MM/GBSA的打分也以ΔG表示;但由于在配体溶剂化处理中存在近似处理,其值通常比实验测得的结合能更负。尽管如此,MM/GBSA仍被视为一种强大的计算方法,可用于对结合于同一蛋白结合位点的分子进行排序。
分子对接可在蛋白构象系综上进行,也可通过能量最小化优化特定结合模式,但上述方法均将蛋白视为刚性结构。而MM/GBSA不仅可在单点进行,还可将蛋白动力学成分纳入计算。通过增加构象采样,可在分子动力学(MD)模拟生成的多条轨迹上计算并平均结合能。
实验SAR分析方法
QSAR模型的成功依赖于配体的叠合方式。尽管在蛋白结构可用时可用于指导叠合,但QSAR计算本身纯属基于配体的计算,不涉及蛋白结构。由于QSAR模型具有统计性质,除需准确反映配体的生物学相关结合模式外,用于训练和测试模型的配体选择也至关重要。
作为指导原则,对于3D-QSAR11模型,建议数据集包含50个以上的配体,且活性范围至少覆盖3个对数单位。虽然构建3D-Field QSAR模型所用化合物数量无上限,但超过200个化合物后收益有限。尽管QSAR模型通常用于理解特定系列内的SAR,但3D-Field QSAR通过XED力场7生成的场点进行参数化,因此可将结合于同一蛋白结合位点的不同化学系列配体纳入同一模型进行研究。
交叉验证决定系数(q2)反映QSAR模型的稳健性,其值介于0(实验与预测活性无相关性)和1(完全相关)之间。通常,若q2 \(>\) 0.4,且所设计配体落在建模数据集范围内,则该QSAR模型可用于指导合成的优先级排序。
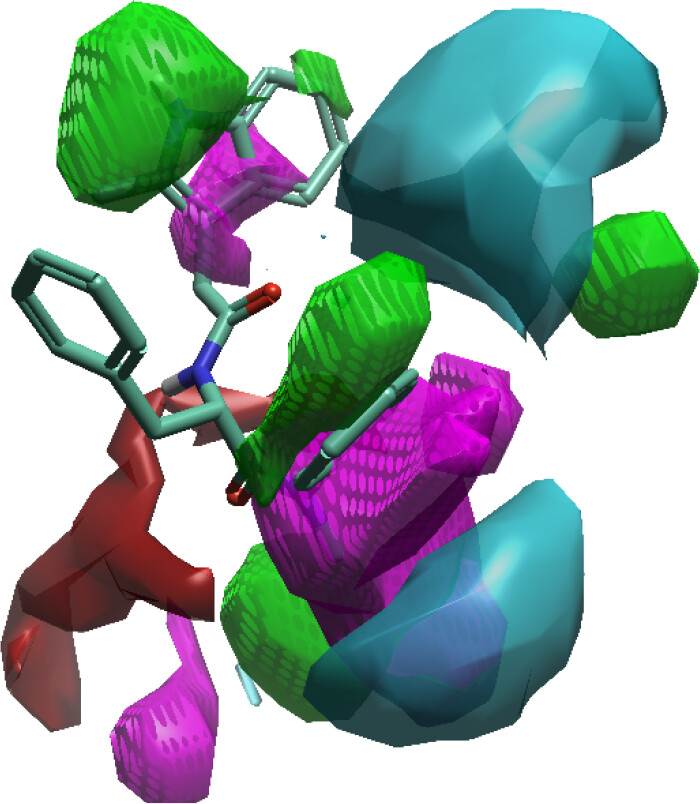
图2. HIV-CA抑制剂(PP-74)的Activity Atlas QSAR模型,突出显示了静电有利(蓝色)、疏水有利(绿色)和 疏水不利(紫色)区域。
利用Activity Atlas,可基于实验数据集生成图像以可视化分析SAR模式。与EC图像不同(其表面颜色基于每个配体–蛋白相互作用生成),HIV-CA 的Activity Atlas图像(图2)12所用的活性悬崖(activity cliff)概括基于整个数据集中的所有配体生成,特定配体仅用于提供上下文。
所有QSAR模型的主要局限在于其仅能提供数据集中包含的信息。例如,QSAR无法预测将配体延伸至此前未探索区域的效果。同样,若某取代基在数据集中所有配体中均存在,则QSAR无法预测该位置变化的影响。因此,在选择QSAR模型化合物时,应尽量包含那些可以覆盖所有感兴趣区域的配体。
结合能的精确预测
自由能微扰(Free Energy Perturbation, FEP)13 已成为计算配体–蛋白复合物结合自由能的金标准13。将FEP应用于项目需满足若干系统要求:首先需要一个合适的配体–蛋白复合物结构作为起点;其次,必须对配体在蛋白环境中的结合模式具有高度确信度,才能对FEP计算结果有信心。
与QSAR类似,FEP也需要实验结合或活性数据用于模型验证。但与QSAR建模需50个以上配体不同,FEP基准研究通常只需约15个配体的数据集。不过,在应用FEP时,仍希望测得的活性具有至少3个对数单位的动态范围。
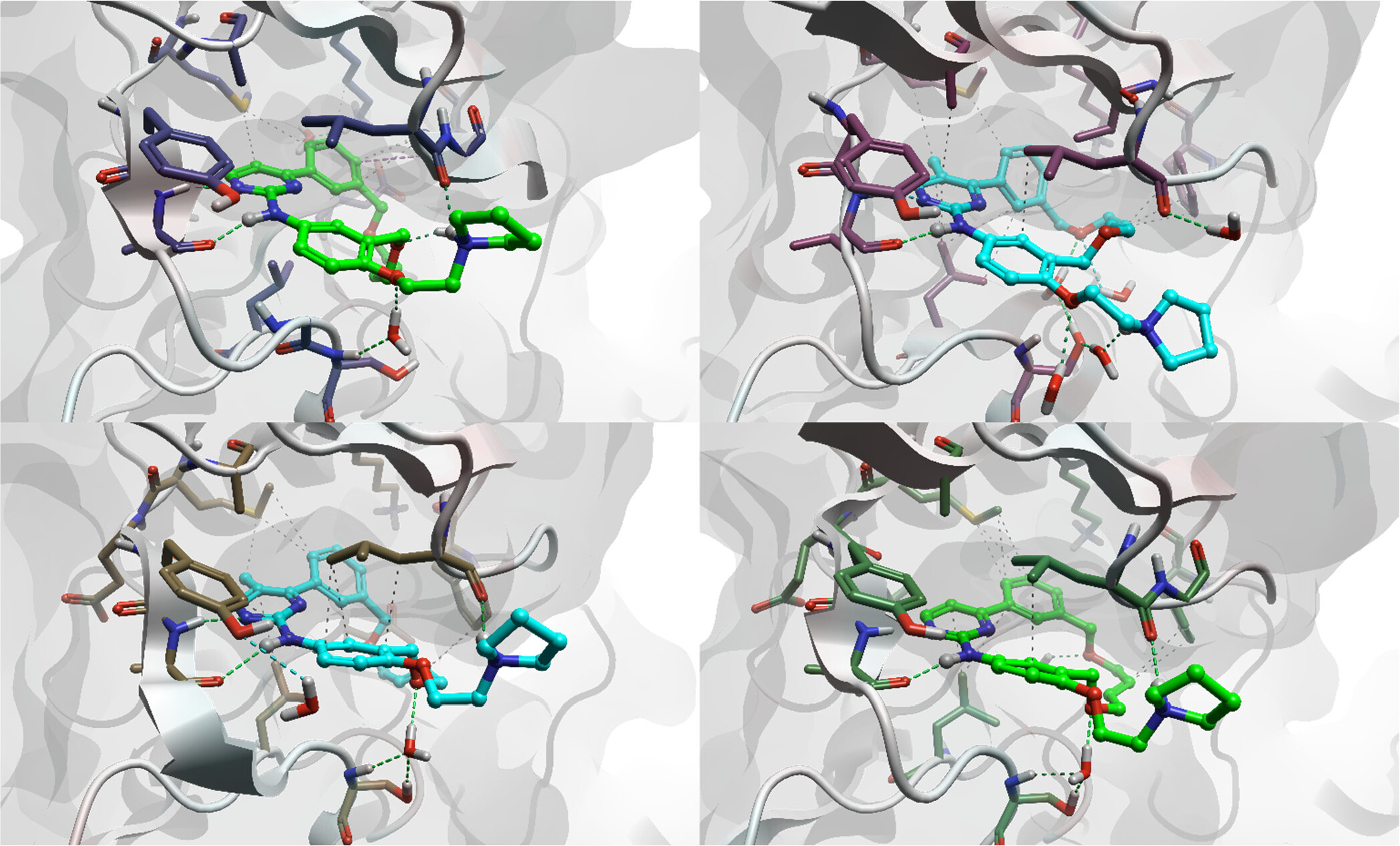
图3. 在pacritinib(绿色)与其5-甲基嘧啶类似物(青色)之间的FEP转化中,JAK2-配体复合物结构的始态(λ = 0)和终态(λ = 1)。
在准备蛋白和配体时需格外谨慎,尤其是体系中某些官能团涉及电荷和互变异构体时。FEP计算将体系准备阶段的初步MD模拟作为必要组成部分,以反映体系的动态特性。水分子采样同样关键,因此建议在体系准备和计算本身中均纳入水分子采样。
FEP计算的复杂性使其计算成本高昂,通常需GPU资源,且一般无法在本地计算机系统上运行。
有两种FEP策略可用于体系。第一种是相对FEP,其中配体通过非物理的“炼金术”路径逐步转化为结构相关的类似物,如图314所示的大环JAK抑制剂配体结合能的相对FEP计算。
在相对FEP中,计算两个配体之间的结合能差值(ΔΔG)。根据转化性质、起始配体–蛋白体系的质量及具体研究的体系,相对FEP计算可非常精确,误差可小于1 kcal/mol(与实验值相比)。
第二种FEP策略是绝对FEP15,其中配体被逐步转化,直至其不再与蛋白环境相互作用。这通过逐步关闭其电荷和范德华参数实现。该过程完成后,即可确定该配体与靶蛋白的结合能。在绝对FEP计算中,计算得到的ΔG可与实验结合能良好相关。
通常,绝对FEP的准确性低于相对FEP。因此,建议使用相对FEP优化现有配体系列,而将绝对FEP保留用于新化学骨架的识别,例如在骨架跃迁(scaffold hopping)中进行优先级排序。
FEP的主要优势之一在于其基于配体与蛋白结合的精确计算,而不像QSAR模型那样依赖统计方法,因此FEP可用于预测超出基准研究探索空间的配体结合能。例如,若基准研究中使用的参数适用于生产级计算,则可预测未探索基团修饰对配体的影响。
选择正确的策略
毫无疑问,计算工具可提升药物发现全过程的效率,挑战在于选择符合项目需求的合适策略。该决策可基于若干因素,例如:
- 可用信息(晶体结构、已知蛋白结合位点/配体构象、实验SAR);
- 项目目标(优化现有系列、识别新化学类型);
- 团队/机构的专业知识与硬件条件(是否有专职计算专家、是否有GPU集群、是否需外包以引入专业知识/硬件)。
| Method | 计算要求 | 得分解读 | 采样方式 | 相对预测能力 | 计算成本 |
|---|---|---|---|---|---|
| Docking | 蛋白质3D结构信息 与配体2D信息 |
ΔG值越负,打分越优 | 单点 | ★ | $ |
| EC | 蛋白质3D结构信息 与配体3D结合构象 |
打分范围[-1,1] -1: 完全冲突 1:完美互补 可视化参见图1 |
单点 | ★ | $ |
| MM/GBSA | 蛋白质3D结构信息与配体3D结合构象 | ΔG值越负,打分越优 | 单点 | ★★ | $ |
| 3D field QSAR | \(>\)50个3D叠合配体,活性范围覆盖3个数量级 | 以实验数据 格式呈现(如pIC50) |
单点 | ★★★ | $$ |
| Activity Atlas | \(>\)50个3D叠合配体,活性范围覆盖3个数量级 | 不适用 | 单点 | 不适用 | $ |
| Relative FEP | 建议\(>\)15个相关3D叠合配体(活性范围3个数量级)及配体-蛋白质复合物结构 | ΔΔG值越负,结合亲和力越优,可通过参考配体转换为pIC50 | 分子动力学 | ★★★★★ | $$$ |
| Absolute FEP | 非相关3D配体及合适晶体结构 | ΔG值越负,结合亲和力越优,可直接转换为pIC50 | 分子动力学 | ★★★★ | $$$$$ |
在项目早期,实验数据有限时,分子对接和EC等技术可为团队提供一种快速且经济的方法,将配体优先级排序至可管理的实验处理水平。随着更多数据积累,定量与定性QSAR方法(如3D-QSAR和Activity Atlas)可高效探索SAR模式,并提升优先级排序的信心,因为这些模型已基于项目的实验SAR进行训练。
随着项目推进,配体设计日益复杂,化合物需基于多个(常相互冲突的)参数进行优化。此时,FEP等计算密集型技术可在开展昂贵的合成及多指标评估(包括体外及后续体内研究)前,显著提升设计合理性的置信度。
为应对日益复杂方法所需的高昂计算成本,可将AI/ML方法整合至主动学习工作流中,例如结合FEP的高精度与QSAR的低计算需求以提升效率16 。据估计,此类主动学习工作流的计算时间不到单独使用FEP的20%。
在正确阶段选择合适的方法可显著提升项目效率。能够优先选择出最优设计候选物进行昂贵的实验评估(如合成与生物表征),不仅能降低整体项目成本,还可缩短到达关键决策节点所需的时间。
随着新型计算方法不断涌现且相关成本持续下降,计算方法在配体排序与结合亲和力预测方面将使更多药物发现流程可在实验前进行计算,从而改善项目成果。


