
从晶体学意外到理性设计:通过模拟硫酸根介导的氢键网络重现补体因子B抑制剂的活性优化
摘要:本文概述了诺华公司开发的首个获批补体因子(Factor B, FB)抑制剂Iptacopan的先导化合物发现历程。研究团队在优化吲哚-哌啶骨架衍生物过程中,通过解析化合物29与FB的共晶结构(PDB: 6QSX),意外发现结晶缓冲液中的硫酸根离子介导了配体与蛋白活性口袋(Arg192、Val218、Asn220B)之间的氢键网络。受此启发,结合结构叠合分析,团队在苯基哌啶对位引入极性取代基以模拟该相互作用。其中,羧酸取代的化合物35展现出显著提升的FB抑制活性(IC₅₀ = 33 nM,较前体提升近100倍),其共晶结构(PDB 6T8V)证实羧酸基团精准复现了硫酸根的氢键模式。
补体系统是人体先天免疫防御体系的重要组成部分,不仅在抵御外源性病原体(如细菌和寄生虫)感染中发挥关键作用,还作为连接先天免疫与适应性免疫的桥梁。该系统由一系列血浆蛋白构成,包括可溶性蛋白、膜结合蛋白及补体受体,主要由肝脏合成或在细胞表面表达,并在血浆、组织液及细胞微环境中行使功能。补体系统的激活主要通过三条经典通路实现:经典通路(classical pathway, CP)、凝集素通路(lectin pathway, LP)以及旁路通路(alternative pathway, AP)。
在健康个体的稳态生理条件下,AP通路维持在低水平的基础活化状态,以持续监测潜在的病原体入侵。此时,补体成分可识别并结合至凋亡细胞表面,但其激活受到严格调控,仅限于清除凋亡细胞,而不引发进一步的炎症或适应性免疫应答。然而,在病原体感染等病理状态下,补体系统被显著激活,通过诱导炎症反应、调理作用及促进吞噬等方式清除病原体,并最终桥接并激活适应性免疫。值得注意的是,补体系统的功能不足或过度激活均可能对机体造成损害,并与多种感染性及非感染性疾病(如自身免疫病、慢性炎症、血栓性微血管病、移植排斥反应及肿瘤等)的易感性密切相关。
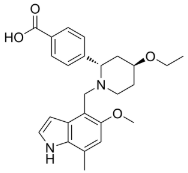
图1. Iptacopan (LNP023)的化学结构式
补体因子B(Factor B, FB)是AP通路中的关键丝氨酸蛋白酶。选择性抑制FB活性可有效阻断AP通路的级联放大,同时避免干扰CP和LP通路,从而在抑制补体过度活化的同时保留其他通路介导的宿主防御功能,降低感染风险。诺华公司开发的LNP023(Iptacopan)是首个获批上市(2023年)1的FB抑制剂,目前已获批用于阵发性睡眠性血红蛋白尿症(PNH)、IgA肾病(IgAN)及C3肾小球病(C3G)等补体介导疾病的治疗。
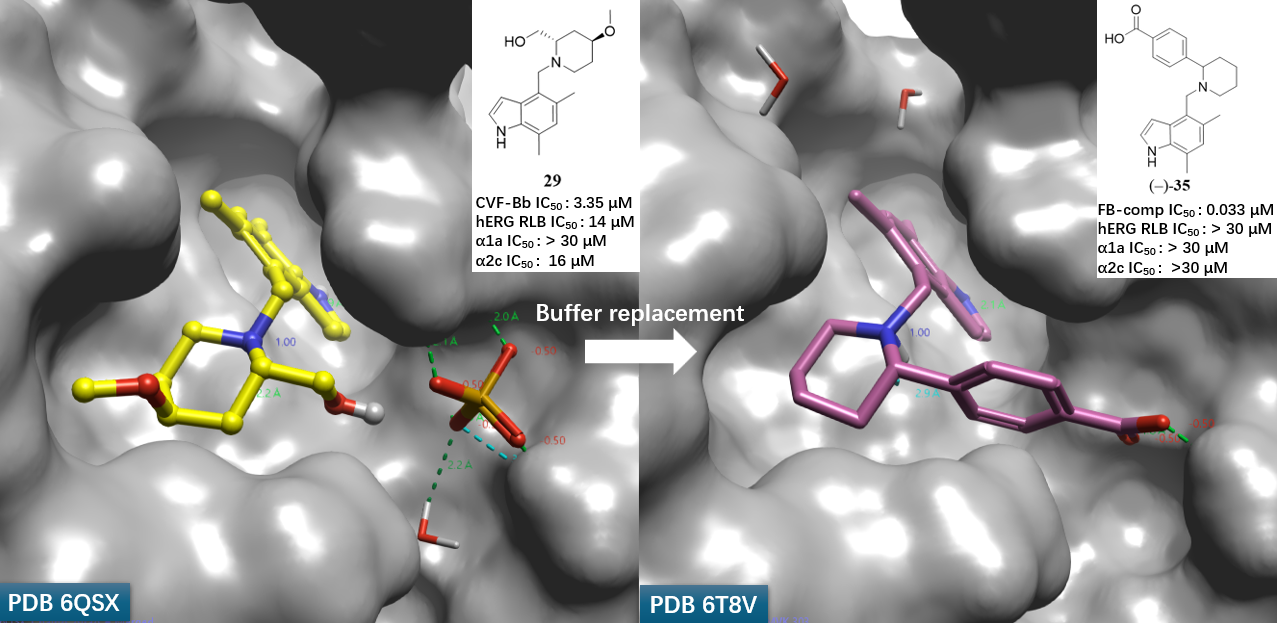
化合物35(图2)的发现是LNP023研发过程中的关键突破,其设计直接源于对前体化合物29(图1)与FB共晶结构的深入解析2。
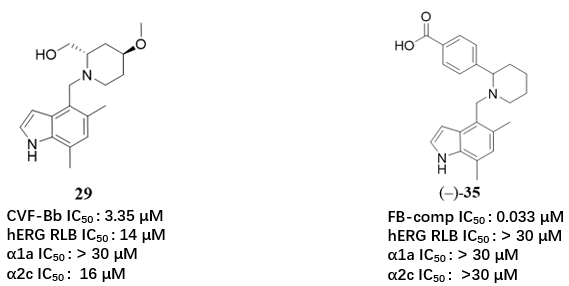
图2. 化合物29与35的化学结构式
在早期优化阶段,研究团队基于吲哚母核与哌啶骨架构建了化合物29(图2)。该分子在FB抑制活性(IC50 ≈ 微摩尔级)及药代动力学性质方面均有所改善。更重要的是,其与FB催化结构域的共晶结构(PDB 6QSX)揭示了一个意外发现:哌啶2位的羟基通过结晶缓冲液中的硫酸根离子形成复杂的氢键网络(图3)。该硫酸根进一步与FB活性口袋中的ARG192、VAL218及ASN220B残基相互作用,提示该区域可作为提升配体结合亲和力的关键位点。
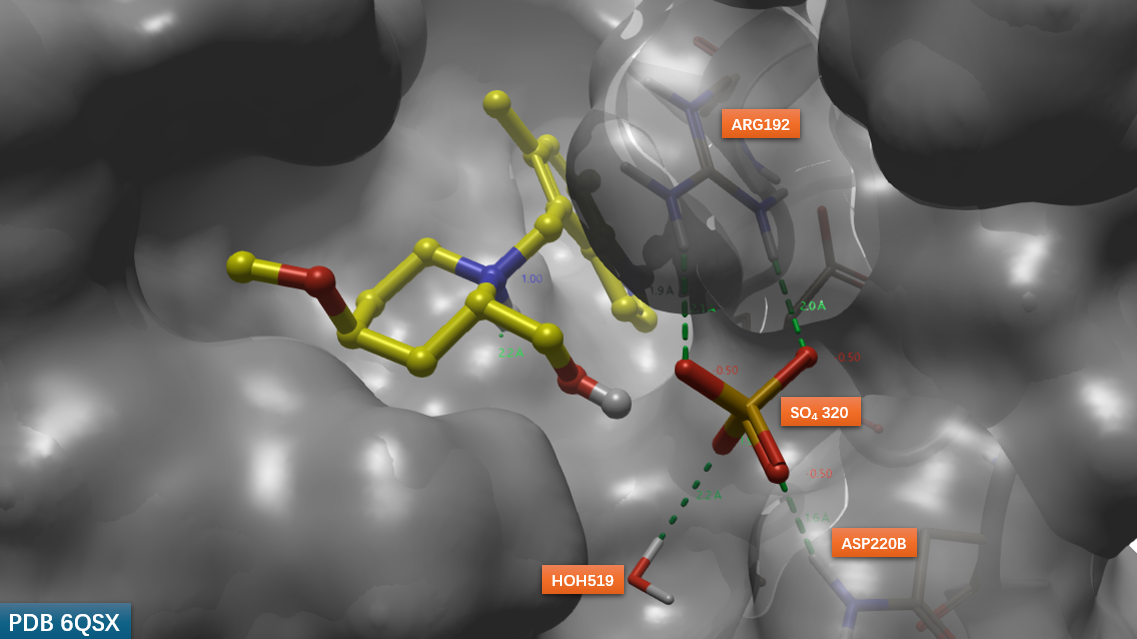
图3. 化合物29与Factor B共晶结构(PDB: 6QSX)显示缓冲液中的硫酸根离子与结合口袋形成氢键网络。黄色球棍:化合物29;灰色表面:Factor B。
这一结构观察为后续分子设计提供了重要启示:若能通过化学修饰模拟硫酸根介导的氢键网络,有望显著增强配体与FB的结合亲和力。
初始尝试直接引入模拟硫酸根的基团未能取得理想效果。然而,通过结构叠合分析,研究团队转向了一种更为高效的策略。将化合物29(PDB 6QSX)与早期苯基哌啶衍生物(S)-19(PDB 6T8W)的共晶结构进行比对(图4),发现(S)-19苯环的对位(para位)恰好占据硫酸根结合的空腔。这一空间匹配提示:在苯环对位引入极性取代基,可能直接模拟硫酸根与蛋白残基的相互作用。
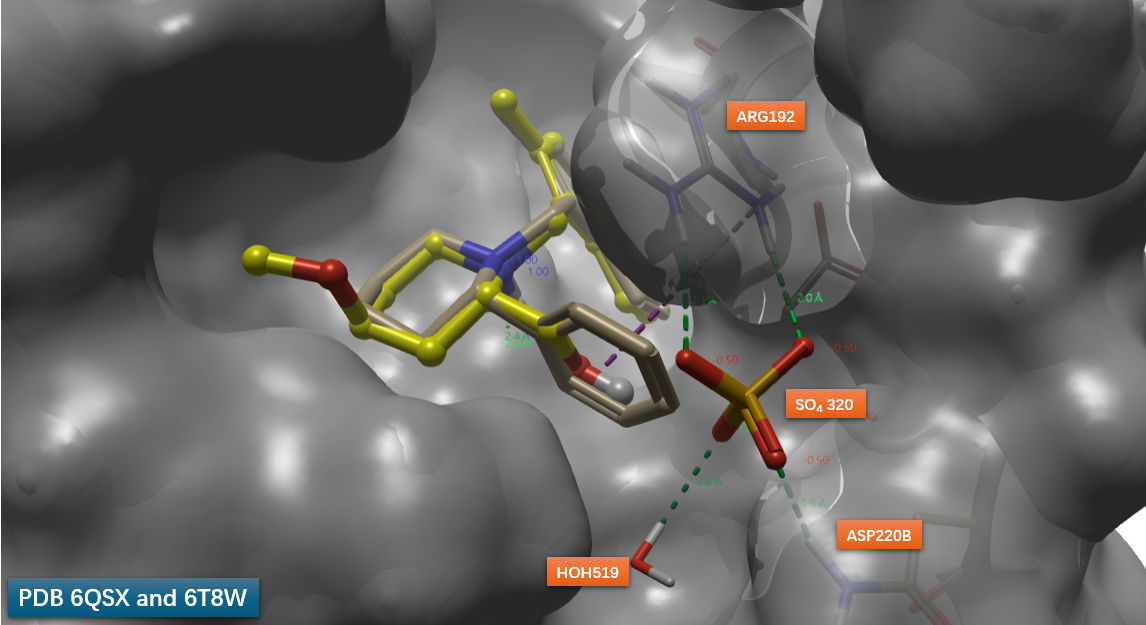
图4. 化合物19、29与Factor B共晶结构PDB 6QSX、6T8W的叠合。其中黄色球棍:化合物29;棍棒:硫酸根;浅棕色棍棒:化合物19;灰色表面:Factor B
基于此假设,团队合成了一系列对位取代的苯基哌啶衍生物。其中,砜基取代(化合物33)使活性提升约4倍;酰胺基取代(化合物34)提升约8倍;而羧酸基取代(化合物35)效果最为显著,其FB抑制活性提升近100倍(IC50由微摩尔级降至33 nM)。选择羧酸基团是基于其强氢键供体/受体能力,可有效模拟硫酸根与蛋白之间的多重氢键相互作用。
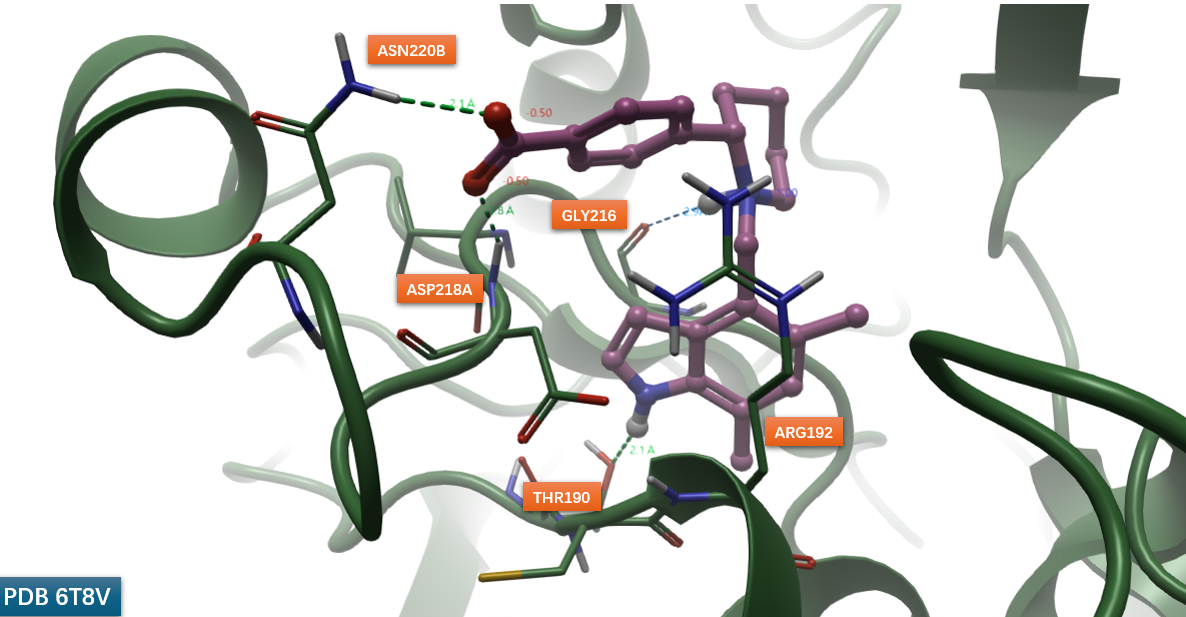
图5. 化合物35与Factor B的共晶结构PDB 6T8V。紫色球棍:化合物35;绿色飘带:Factor B
(-)-35与FB的共晶结构(PDB: 6T8V)验证了上述设计策略:其羧酸基团与Asn220B侧链及Asp218A主链NH形成强氢键(图5),精确复现了硫酸根的结合模式。除显著提升FB抑制活性(IC50 = 33 nM)外,(-)-35还完全消除了对hERG通道及肾上腺素受体(α1a/α2c)的脱靶效应,确立了其作为高质量先导化合物的地位。
综上所述,先导化合物35的发现充分体现了基于结构的药物设计(structure-based drug design, SBDD)与构效关系(SAR)分析在现代药物研发中的核心价值:通过晶体学偶然发现(硫酸根介导的氢键网络)转化为理性分子设计(苯环对位引入羧酸基团),成功突破了活性瓶颈,为后续临床候选物LNP023的诞生奠定了基础。
为评估该设计路径是否可通过计算手段系统性重现,我们采用行业领先的生物电子等排体替换软件 Spark3 开展了回溯性虚拟生物等排体替换实验。具体而言,我们利用 Spark 中的 “Scaffold hopping or R-group Replacement” 模块,旨在验证:能否将结晶缓冲液中作为蛋白–配体氢键网络中介的硫酸根离子,视为可被理性替换的极性功能单元,并通过计算策略指导配体优化?
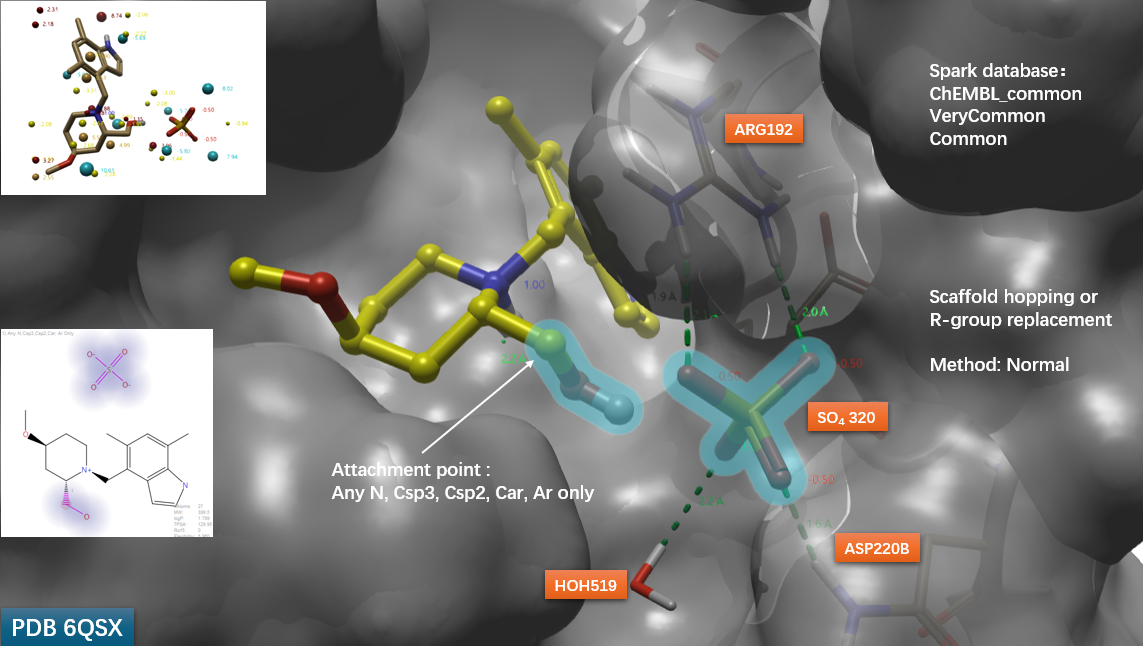
图6. 基于 Spark “Scaffold hopping or R-group Replacement” 模块的替换实验设置。高亮区域包括化合物29(黄色球棍)的哌啶侧链羟甲基与邻近的硫酸根离子,共同定义为替换片段;灰色表面代表 Factor B 蛋白(PDB: 6QSX)。
具体而言,实验以 PDB 6QSX 中的配体29及其邻近硫酸根为参比结构(Reference),将二者组成的高亮部分(图6)指定为被替换区域,并设定附着点(attachment point)的原子类型为:Any N、Csp³、Csp²、Car 与 Ar only。随后,采用标准计算模式(Method: Normal),对 ChEMBL_common、VeryCommon 与 Common 三个子数据库进行虚拟筛选。
Spark 共生成 500 个高打分替换方案。值得注意的是,多个关键已知分子被成功重现:化合物35(即 LNP023 的先导化合物)出现在结果第 #206 位;此外,排名靠前的 #1、#24 与 #94 等结构亦已被 ChEMBL 数据库收录为已知的补体因子B抑制剂(图7)。更重要的是,该实验还产出了大量未见文献报道的新型化学骨架,其计算打分与已知活性分子相当,提示其具有潜在的高亲和力与结构新颖性,值得进行下一步评价,并最终通过合成与体外活性测试验证。
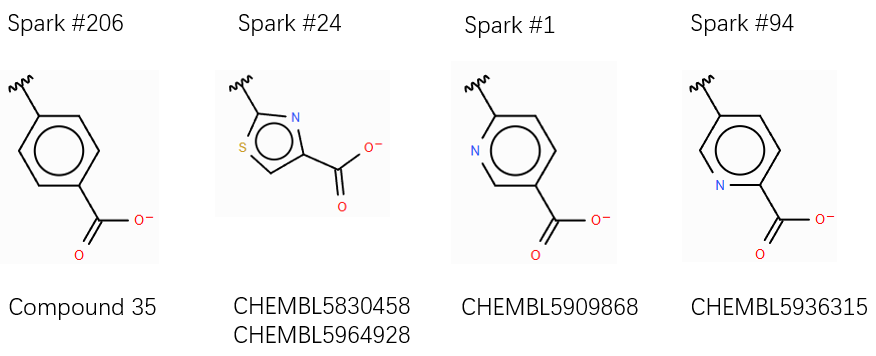
图7. Spark “Scaffold hopping or R-group Replacement” 实验中部分经文献验证的结果示例
该回溯性分析表明,将介导蛋白–配体氢键网络的结晶缓冲液离子(如硫酸根)纳入 Spark 的 “Scaffold hopping or R-group Replacement” 框架,可有效实现对该类关键极性相互作用的理性模拟与替换。该方法不仅成功复现了历史研发中的关键突破,还高效生成了结构多样、高潜力的候选分子,显著加速了基于结构的先导化合物优化进程。这一策略为靶向由离子或水分子介导的蛋白–配体氢键网络的结合口袋,提供了一种可推广的计算设计范式。
文献
- 中国国家药监局药品审评中心官网. Retrieved October 23, 2025, from https://www.cde.org.cn/main/xxgk/listpage/da6efd086c099b7fc949121166f0130c.
- Mainolfi, N. et al. (2020) “Discovery of 4-((2 S ,4 S )-4-Ethoxy-1-((5-methoxy-7-methyl-1 H -indol-4-yl)methyl)piperidin-2-yl)benzoic Acid (LNP023), a Factor B Inhibitor Specifically Designed To Be Applicable to Treating a Diverse Array of Complement Mediated Diseases,” Journal of Medicinal Chemistry, 63(11), pp. 5697–5722. Available at: https://doi.org/10.1021/acs.jmedchem.9b01870.
- Spark. Cresset. https://www.cresset-group.com/software/spark


