
摘要:本文评估了XedeX软件在分子构象搜索中的性能优化。采用OpenEye精心筛选的PDB配体数据集(经清理后含192个配体),比较了XedeX与商业工具Omega的构象生成能力。结果显示,两者整体性能相当:XedeX在精确匹配生物活性构象(RMSD\(<\)0.25Å)时略优(11.6% vs. 9.6%),而Omega在中等RMSD范围(0.75–1.25Å)稍占优势。研究发现,XedeX的性能随分子可旋转键数量增加而显著下降,这与Omega的稳定性形成对比。深入分析揭示,XedeX在特定分子(如1s63)上的失败源于其简化力场(关闭长程静电和吸引性范德华力)忽略了关键分子内相互作用(如阳离子-π作用)。这突显了构象搜索中计算速度与准确性之间的固有权衡,改版的力场虽提升效率,但牺牲了对复杂构象的捕捉能力。
原文:Cresset (May 25th, 2011). Conformation search performance with XedeX.
编译:肖高铿
一两年前,我们开展了一些工作,研究XedeX中的各种选项及其最佳值,以优化构象搜索性能。当然,要做到这一点,你需要定义一个度量标准!文献中的普遍共识似乎是,最佳的度量标准(至少在虚拟筛选的背景下)是取一批PDB配体,从一维或二维输入中为其生成构象,然后查看这些构象与生物活性构象的接近程度。但这种方法存在几个问题:
- PDB中充满了精修不当和指定不准确的配体。
- 测试结果对你生成构象的数量非常敏感,尽管以1°为增量的穷举系统搜索必然会找到正确答案,但这种方法并不实用。
- 使用RMSD作为度量标准也存在问题,因为它对分子大小敏感。对于片段分子来说,2.0Å的RMS误差非常糟糕,而对于四肽分子来说,这个误差实际上还算可以接受。
尽管如此,这仍然是目前最好的建议,因此我们采用了这种方法。最初的工作相当冗长复杂,所以我们可能会在后续的文章中详细描述。简而言之,通过调整选项,我们得到了一套新的“默认”设置,其性能明显优于原始设置,因此FieldScreen已升级使用这套新设置。
OpenEye最近发表的一篇论文提出了一个用于此类分析的新数据集,其中蛋白质结构经过精心筛选,以消除将分子与不良参考构象进行比较的问题。因此,我们认为有必要重新审视我们的分析,以确保我们之前没有被有问题的数据集误导!在整理数据集时,我们遇到了一些问题:数据集中有些配体我们认为不合适,因为它们是共价结合的;另外还有一些配体,原作者的结构与我们的解读不同,我们必须查阅原始论文来确定配体的真实结构。解决这些问题后,数据集的大小从197个减少到192个,其中约20个配体与原始数据集相比进行了修改。
总体而言,我们的性能与Omega的性能相当接近:
| Method | RMSD (Å) | ||||||
|---|---|---|---|---|---|---|---|
| \(<\)0.25 | \(<\)0.5 | \(<\)0.75 | \(<\)1.0 | \(<\)1.25 | \(<\)1.5 | \(<\)2.0 | |
| Omega(200 confs) | 9.6 | 50 | 71.3 | 83.0 | 87.2 | 92.6 | 96.8 |
| XedeX(200 confs) | 11.6 | 52 | 68.0 | 81.8 | 88.5 | 93.6 | 97.6 |
表格中的数字表示在给定RMSD阈值范围内的正确率百分比——我们将其表示为百分比而非绝对数值,因为XedeX的结果是使用不同随机数种子进行5次重复运行的平均值(这是一种随机方法)。两个程序得出的相对数值很有意思:XedeX在完全正确(RMSD\(<\)0.25)的分子数量上更多,Omega在中等范围表现稍优,而在"错误"区间两端的方法表现大致相当。不过,这些结果需要谨慎看待,因为两组数据集并非完全等效(由于前文提及的问题)。
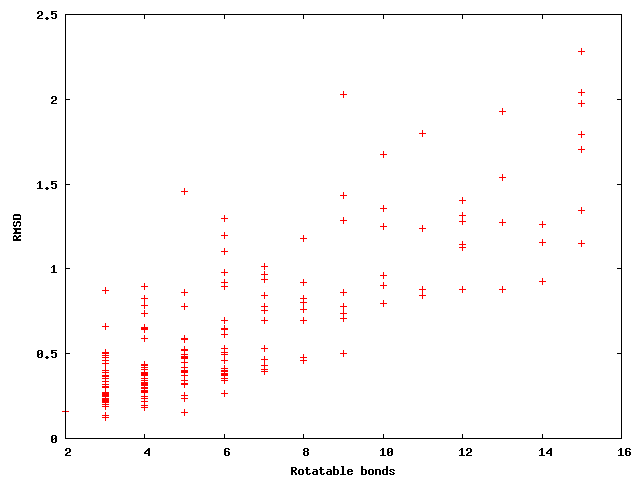
通过分析XedeX在这个数据集上的表现,我们还发现了一些有趣的现象。首先,找到生物活性构象的几率会随着可旋转键数量的增加而下降:
或者,如果您愿意,可以使用场相似性度量而非RMSD来呈现相同的数据:
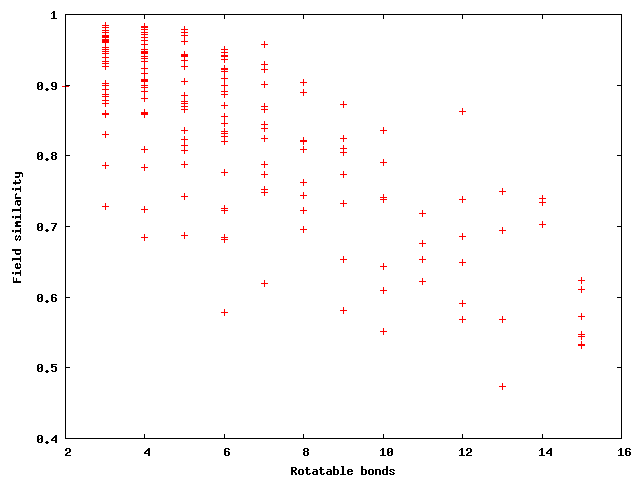
你可能会说这并不奇怪,因为分子的可旋转键越多,其构象空间就越大,找到接近生物活性构象的可能性就越低。然而,OpenEye的私下交流显示,这一规律对Omega并不适用。这相当令人惊讶:我能想到的唯一解释是,他们基于规则的算法既有可能严重偏离某些小分子的生物活性构象(如果这些构象的扭转角恰好不在扭转规则数据库中),又可能更准确地获取某些大分子的构象(如果这些大分子的所有扭转角都非常接近扭转角数据库中的值,那么你就有机会几乎精确地生成生物活性构象,而这对随机搜索方法来说要困难得多)。
我们观察的第二个重点是XedeX在哪些分子上表现不佳——即那些RMSD值很高但可旋转键数量较少的分子。其中特别突出的是1s63这个分子,它只有5个可旋转键,但我们只能达到距离生物活性构象约1.45Å的RMSD值。观察其生物活性构象后,问题变得一目了然:
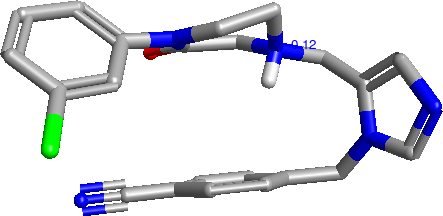
分子通过一个精美的阳离子-π相互作用保持折叠构象。XED力场确实能非常精准地处理阳离子-π相互作用,因此您可能认为这很容易实现。然而,标准XedeX条件使用的是改版的XED力场,其中关闭了吸引性的长程静电作用和范德华力(作为溶剂化模型的快速近似),这带来的副作用是忽略了分子内氢键。Omega显然也采用了类似方法,对该分子的RMSD值为1.7Å。如果使用完整力场运行XedeX,则能得到正确结果(RMSD为0.41Å)。这就是使用改版力场需要做出的权衡——对大多数分子能得到更优结果,但对存在分子内氢键的构象就会失效。采用”真正”的溶剂化模型可能解决这个问题,但计算速度会过于缓慢。


