
摘要:在缺乏靶标蛋白高分辨率晶体结构或已知生物活性构象的情况下,如何准确预测多个结构迥异但功能相似的小分子配体的生物活性构象(bioactive conformation)是基于配体药物设计的核心挑战。本研究以二肽基肽酶-4抑制剂(DPP-IV inhibitors)为模型体系,利用 Cresset 药物设计平台 Flare 的FieldTemplater 工具,通过整合四个结构多样、活性模式相似的先导化合物,尝试构建可靠的场点模板(Field Template),用于预测其潜在的生物活性构象与关键相互作用特征。
作者: Jessica Plescia/September 22, 2025
编译: 肖高铿
引言
在早期药物发现阶段,关于生物靶标的可用信息往往仅限于其对少数特定配体的活性数据。由于缺乏三维结构模型(或任何其他结构信息),配体的活性构象(即结合态构象)无法确定。此类结构模型的缺失可能由多种因素导致,例如靶蛋白结构过于不稳定而难以实现成功结晶。在此情况下,可采用基于配体的计算方法来替代基于结构的方法。相关策略包括建立物理性质与生物活性之间关系的模型,如2D/3D-定量构效关系(2D/3D-QSAR)或药效团模型。Cresset公司开发的Flare™平台中的FieldTemplater™是此类方法的典型代表。该方法首先对少量已知活性配体结构(可从2D结构出发)进行构象搜索,随后尝试将所得的3D构象通过一种类似于“链接网络”的方式相互叠合1。尽管未依赖于靶标蛋白的晶体结构,该流程仍可生成一系列具有良好结合模式预测能力的配体模板集合。这些模板可作为参考框架,用于指导新配体设计或用于对虚拟化合物库中分子的三维构象进行系统性叠合分析2。当然,此类实验的前提假设是所有配体均结合于同一结合位点:若配体结合于不同位点,则无法建立可靠的结合位点假设,因为该基本前提不成立。同样重要的是,参考配体的分子尺寸应相近,因显著的分子量差异可能导致对生物活性构象的预测出现偏差,从而降低模型的准确性。
二肽基肽酶IV(DPPIV)——一个典型的示范案例
为展示FieldTemplater™方法的实用性,本文以二肽基肽酶IV(Dipeptidyl Peptidase IV, DPPIV)为例进行说明。DPPIV是一种丝氨酸蛋白酶,在糖尿病和癌症等疾病中具有重要生物学意义。在正常细胞中,DPPIV的主要功能之一是调控代谢与葡萄糖稳态,例如通过切割胰高血糖素样肽-1(Glucagon-like Peptide-1, GLP-1)——一种参与食欲抑制与胰岛素分泌的激素——来实现其生理调节作用3。美国食品药品监督管理局(FDA)已批准的DPPIV抑制剂,即“gliptins”类药物,最初设计用于2型糖尿病患者的血糖控制治疗4。在癌症领域,DPPIV亦被证实参与肿瘤进展与转移过程,其机制涉及对免疫应答的调控以及对细胞黏附功能的影响5。因此,DPPIV在多种病理生理过程中的多重角色凸显了其作为潜在治疗靶标的显著价值。图1展示了四种已知的强效DPPIV抑制剂的 2D 与 3D 结构,为后续基于配体的结合模式预测提供了关键参考。
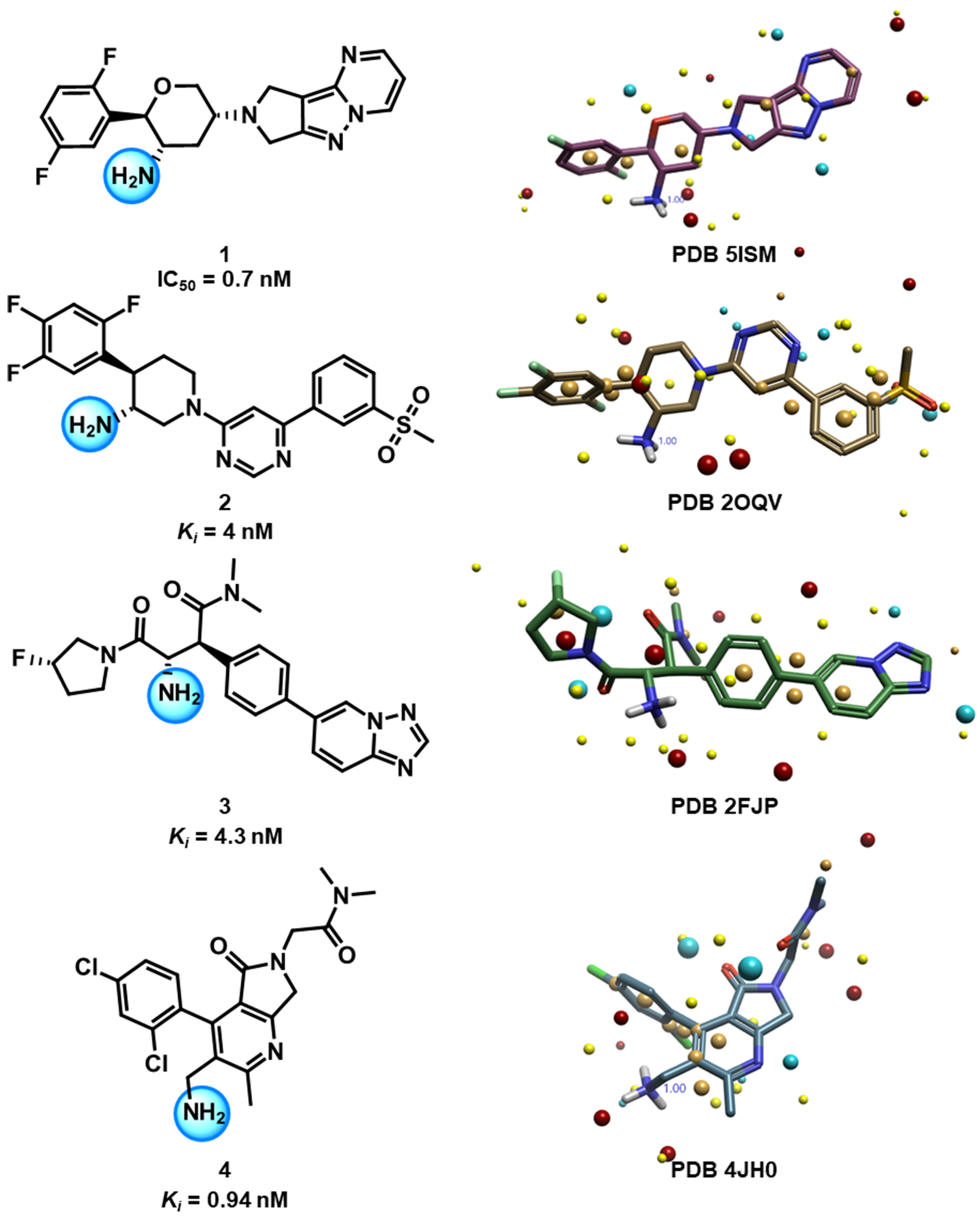
图1. DPPIV的低纳摩尔级结合物及其活性数据。左图:二维结构,突出显示游离氨基基团;右图:三维晶体结构,展示Cresset场点(Field Points)分布6, 已根据生理pH值7进行调整以增强生物学相关性。
如图1所示,四种抑制剂具有显著不同的化学结构,但其分子尺寸相近(以化合物1为参照计算的Morgan二维相似性值范围为0.39–0.57)。仅基于二维化学结构分析,这四个化合物均含有位于“铰链样”骨架上的氮杂环结构,该结构在空间上呈现负电性特征,从而与具有正电性特征的蛋白质结合口袋形成静电互补。其芳香基团很可能与活性位点中的芳香族氨基酸残基发生芳香相互作用。然而,这四个化合物共有的唯一官能团是游离胺/铵基。值得注意的是,在蛋白质结合位点中,该铵基可与两个谷氨酸残基形成氢键及盐桥相互作用(以化合物1为例,见下图2)。在FieldTemplater实验中,我们假设这四化个合物在蛋白结合过程中均形成高度相似的相互作用模式,尤其是与铵基相关的氢键网络和盐桥。鉴于此,在本案例中聚焦于单一官能团(即铵基)的约束条件,相较于施加更复杂的整体静电场约束更为简便且高效。实验目标为:以配体的二维结构为起点,通过生成的模板结构与已叠合的晶体结构进行比对分析,从而实现对结合模式的系统性评估与优化。
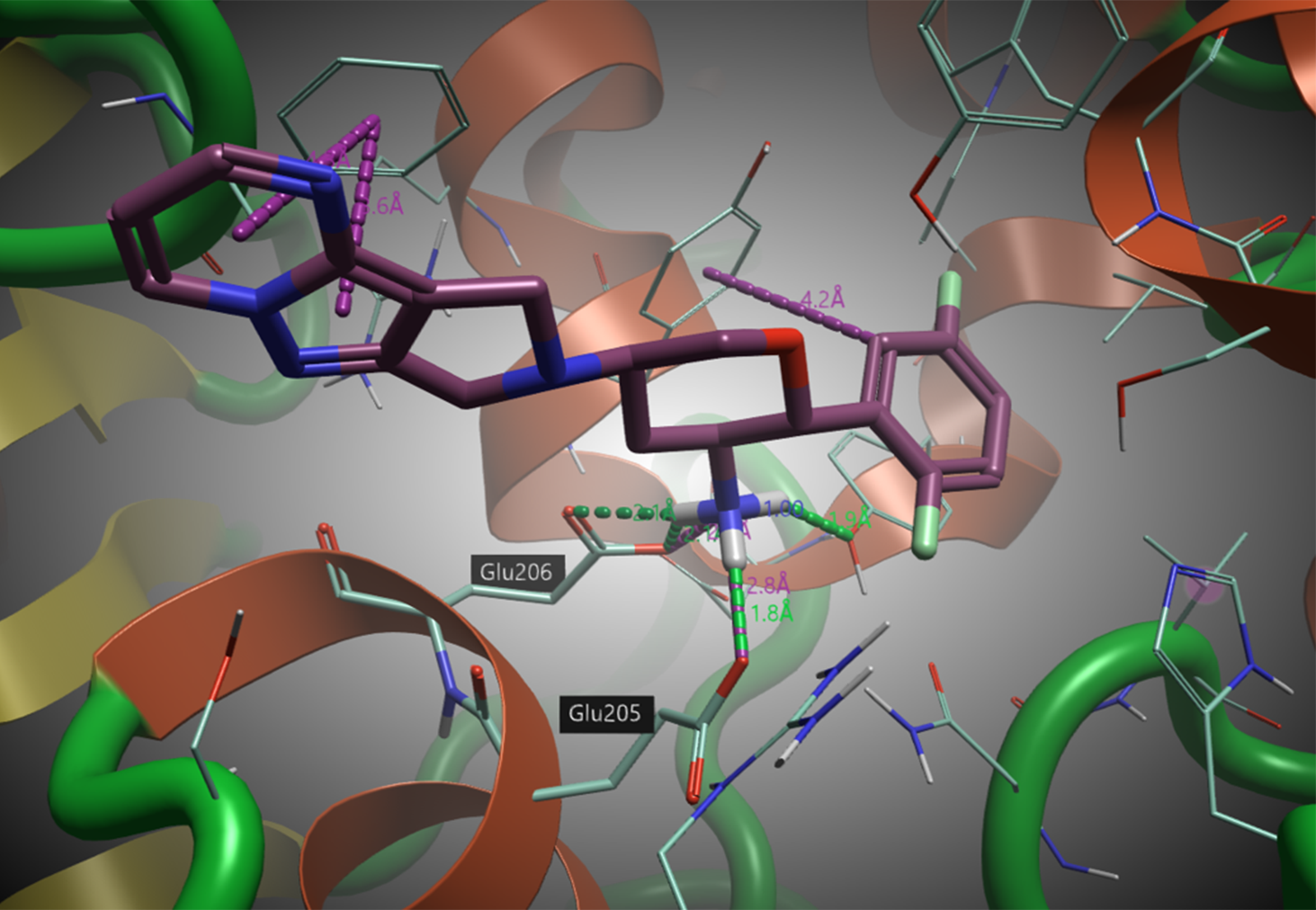
图2. 化合物1与DPPIV(PDB code:5ISM)结合的结构可视化结果,展示于Flare平台中。谷氨酸残基205和206已标注。
方法
从四个化学结构差异显著的抑制剂中获取模板
首先,将配体以SMILES字符串形式导入Flare软件,并采用生理质子化态进行建模,即胺基均被质子化为铵基(ammonium)。随后,利用Cresset XED力场对体系进行能量最小化处理(尽管此步骤并非必需)。接着,使用FieldTemplater工具在默认参数条件下运行,具体设置包括:最大构象数为100,Sim打分中形状相似性贡献权重为50%,每对化合物间最大比较次数为100,每对化合物间最大Δ打分阈值为0.10(见图3)。根据贡献化合物间的平均Sim值进行排序,前四个最优模板在结果表格中被高亮显示,并在三维视图中呈现。
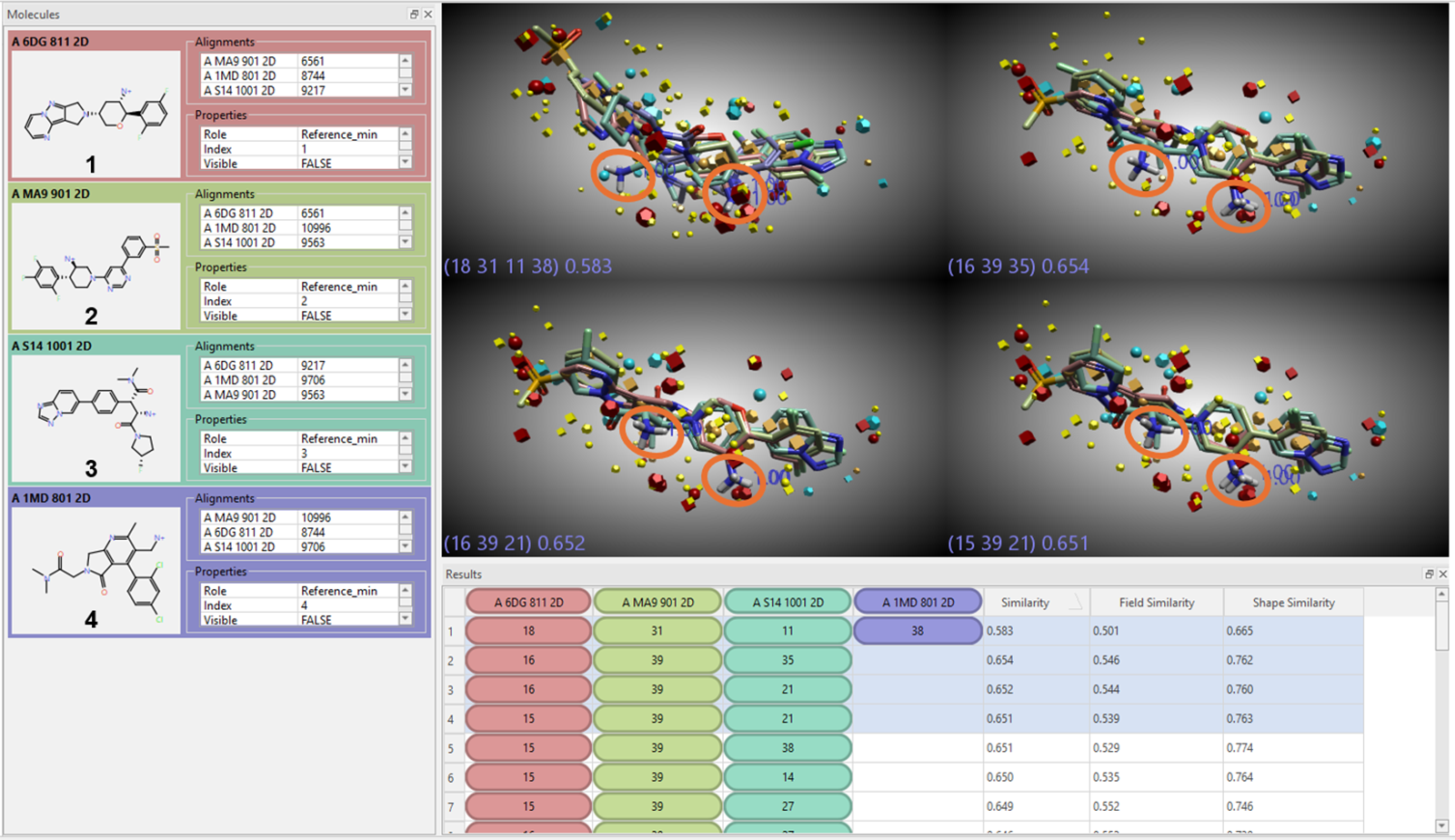
图3. 第一次FieldTemplater实验(常规设置)中生成的前四个模板结构。游离铵基以橙色圆圈高亮显示。
显然,前四个最优模板并未准确反映晶体结构中的配体结合构象;铵基之间极少出现空间叠合。此外,蛋白质结合位点内可能呈现更强正电性或负电性的区域缺乏明确共识——场点(field point)聚类分布杂乱无章,未形成具有清晰正负电性特征的集中区域。上述两点观察结果可能是关键信号,提示当前生成的模板未能真实表征蛋白质结合口袋的电性环境。为获得更具生物学合理性的模板,可引入成对约束(pairwise constraints)(见图4)。成对约束通过强制算法优先选择特定原子间距离在预设阈值内的构象排列,从而引导模板生成过程。本案例中,设定各化合物中胺基氮原子之间的距离应保持在1 Å以内(采用默认设置),以确保铵基在空间上高度协同定位,进而强化其与蛋白活性位点中关键酸性残基(如谷氨酸)形成氢键及盐桥相互作用的潜力。该策略有助于生成更符合实际结合模式的、具有明确电性特征和空间取向一致性的模板结构。
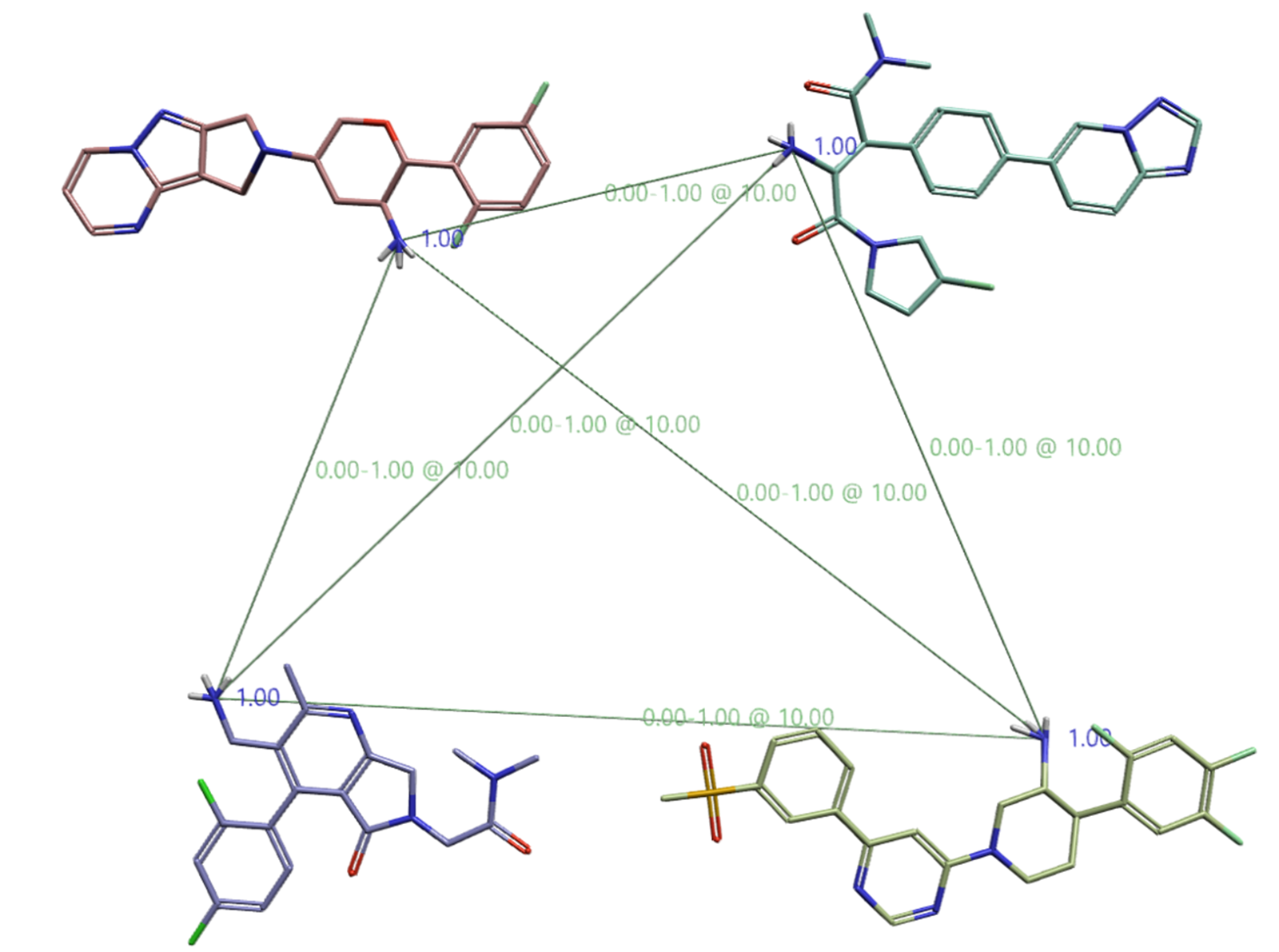
图4. 成对约束条件的设置。在所有铵基对之间默认设置了1.00 Å的约束条件。成对约束窗口可通过FieldTemplater窗口中的“主页”(Home)选项卡打开。绿色标签表示距离约束容差范围为0.00–1.00 Å,并关联一个任意设定的约束惩罚值10.00,该值用于惩罚那些不满足约束条件的构象。
在设定上述约束条件后,采用默认的构象搜索(conformation hunt)、分子叠合(alignment)及模板生成(templating)参数重新运行FieldTemplater实验,结果如图5所示。
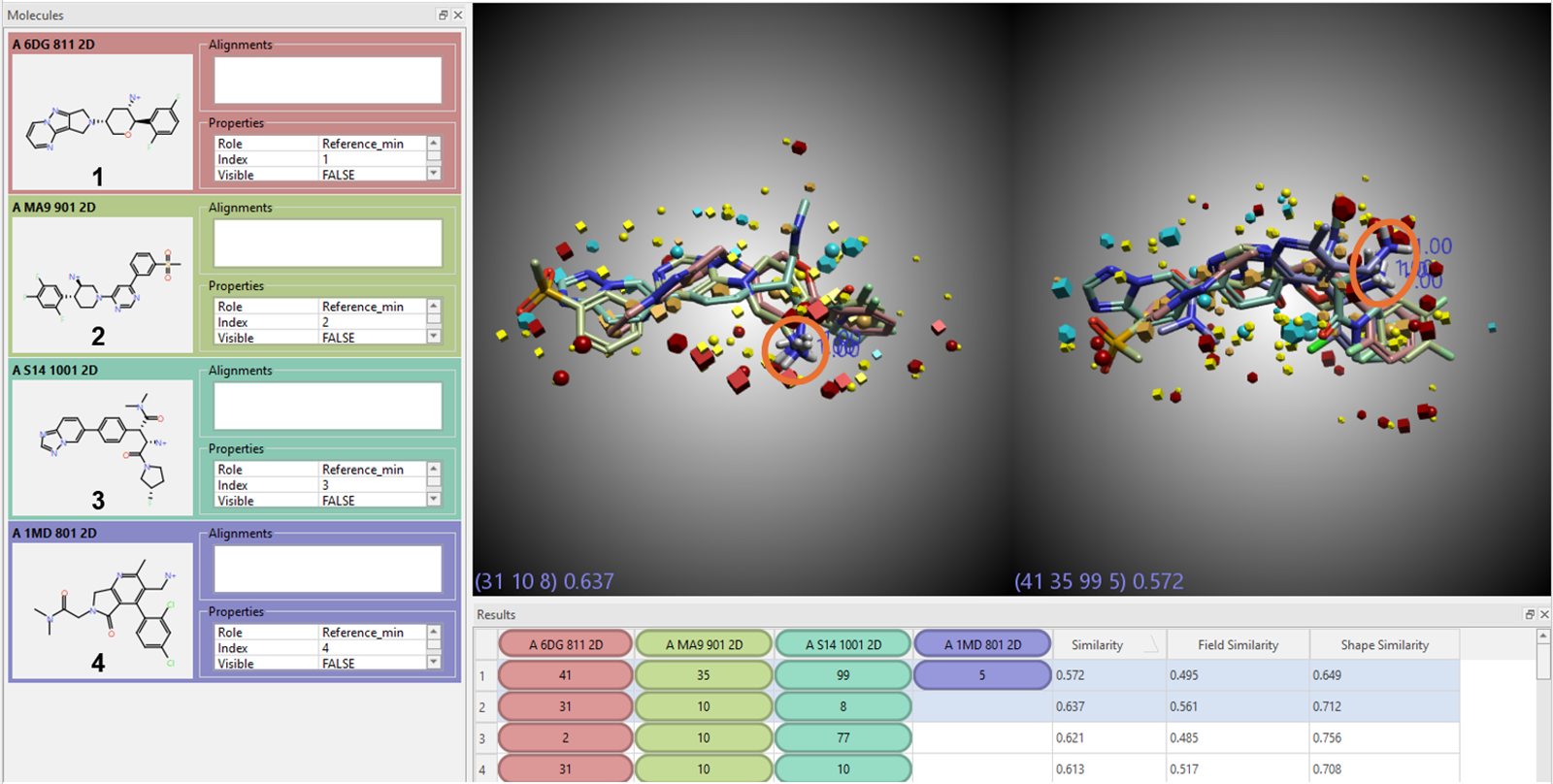
图5. 在默认设置下结合游离铵基成对约束条件所获得的前两个最优模板(结果表格中已高亮,并在三维视图中展示)。
在实验结果中,输出模板仅包含一个由全部四个化合物共同构成的模板,其余模板均基于其中三个化合物构建。观察前两个最优模板可见,二者均将成对约束(即自由铵基之间的空间距离限制)作为分子叠合的核心焦点。然而,两者的相似性打分存在显著差异:包含全部四个配体的最佳模板(Template 1)相似性打分为0.572,而仅包含其中三个化合物的次优模板(Template 2)相似性打分更高,达0.637。
在缺乏配体真实生物活性构象(bioactive conformation)先验信息的前提下,FieldTemplater预测:Template 2 所代表的分子叠合方式更具可能性。进一步分析表明,尽管Template 1整合了全部四个化合物,但其构建依赖于化合物3(A S14 1001)的第99个构象(共100个),该构象能量极高,属于高能态构象,其构象稳定性较差,可能不具备生物学合理性。相比之下,当将化合物4(A 1MD 801)从模板中剔除后,程序可选用其余三个配体的低能构象进行叠合,从而构建出更具能量优势且共识性更强的结合模式。
将上述两个模板分别通过旋转与平移操作,将其与晶体结构(crystal structure)进行叠加比对,结果表明:Template 2 在空间取向上与晶体结构中的实际配体结合模式构象高度一致,其对接结合模式(docking pose)与实验观测的生物活性构象更为接近。该结论在图6中得以直观展示,同时通过RMSD值量化分析(见表1),进一步证实Template 2在空间匹配度上显著优于Template 1。
综上所述,尽管Template 1在化合物覆盖面上更全面,但其依赖高能构象的特性削弱了其可靠性;而Template 2虽仅包含三个化合物,却凭借更高的相似性打分、更低的能量构象以及与晶体结构更强的空间一致性,成为更具可信度和指导意义的模板,为后续的骨架跃迁(scaffold hopping)及先导化合物优化(lead optimization)提供了更可靠的结构基础。
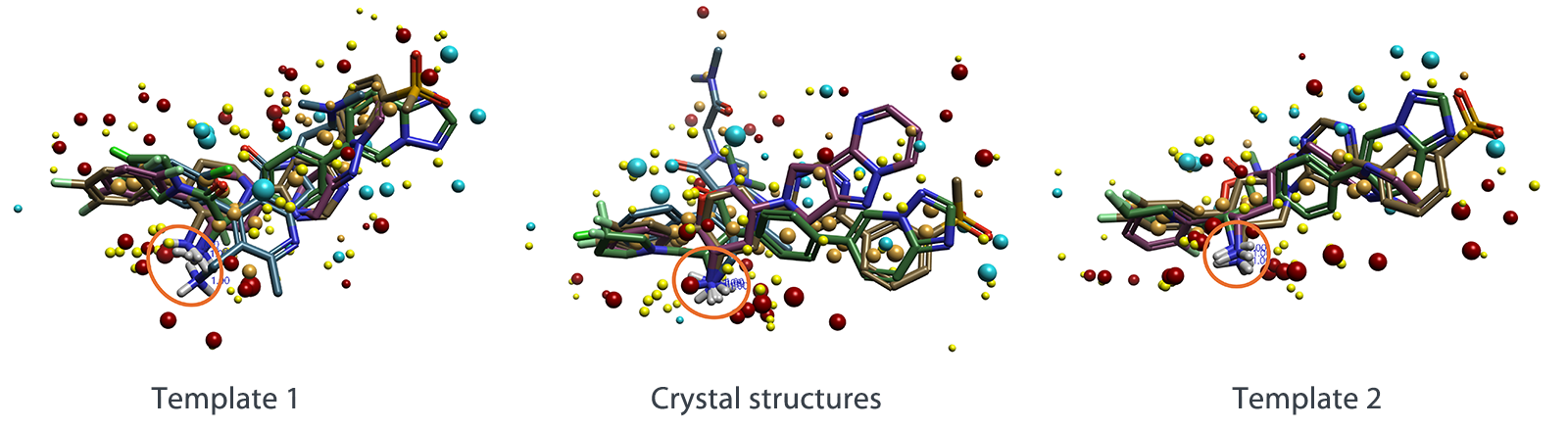
图6. FieldTemplater获得的前两个最优模板结构与晶体结构配体的对比。游离铵基以橙色突出显示。化合物1以紫色展示,化合物2以棕色展示,化合物3以绿色展示,化合物4以蓝色展示。
尽管存在结构上的差异,这些模板仍可作为可接受的参考构象使用。在FieldTemplater实验中,可以通过调整更多参数来尝试优化结果,例如增加每对模板的最大打分差异或每对模板使用的最大叠合数量。此选项规定了最佳打分叠合与任何待使用叠合之间相似性得分的最大差异。此外,还可以提高最小连接密度,这将增加构建此模板时必须使用的最小成对连接比例。然而,实验发现增加得分差异和连接密度并未显著改善实验结果。
表1. 各模板与晶体结构取向之间的距离比较
| Ligand | RMSD to crystal structure (Å) | |
|---|---|---|
| Template 1 | Template 2 | |
| 1 | 2.0 | 2.8 |
| 2 | 4.0 | 1.3 |
| 3 | 3.7 | 2.1 |
| 4 | 5.5 | – |
正如Sim打分所预测,并经与晶体结构的RMSD值证实,模板2在构象和取向上都比模板1更接近晶体结构。同时我们也能观察到,模板2的静电特性与晶体结构更为相似——其下部区域呈现更强的静电正性,而上部区域则带有更多静电负性,且局部还存在一些正电性的分布区域。相比之下,模板1虽然Sim打分较低、能量构象较高,但整体表现出强烈的静电正性,且负电场区域似乎较为分散。值得注意的是,在本次实验中,仅使用四个参考配体中的三个,便能生成更为精确的模板。如需调整模板中化合物的最低数量,可随时修改“模板化”设置。
结论
在本研究中,使用FieldTemplater生成了基于配体的DPPIV结合剂生物活性构象预测。当应用基于一个简单假设的约束条件时——即唯一共同的官能团对这些配体的活性至关重要——结构多样的输入配体给出了可接受的结果。在这些限制条件下,FieldTemplater能够在大约1.3-2.8埃的精度范围内预测它们的生物活性构象。此外,场点集群与晶体结构的场点高度相似。这些结果突显了FieldTemplater在生物靶标结构信息非常有限的情况下的实用性。对于具有共同锚定位点的化合物,包括锌结合基团、共价弹头或如本例中的关键公共官能团,FieldTemplater是一个特别有用的工具。此处生成的打分靠前模板可作为生物活性构象参考,用于配体叠合实验或基于配体的虚拟筛选。
软件试用
想要亲自在您的基于配体项目中尝试FieldTemplater,请立即联系我们以观看Flare的演示。
参考文献
- https://cresset-group.com/software/fieldtemplater
- Chisholm, T. S., Mackey, M., Hunter, C. A. J. Am. Chem. Soc. 2023, 145, 29, 15936–15950. https://pubs.acs.org/doi/10.1021/jacs.3c03749
- Deacon, C. F. Front. Endocrinol. 2019, 10, 80. https://doi.org/10.3389/fendo.2019.00080
- Ikuma, Y., et al. Bioorg. Med. Chem. 2012, 20, 5864–5883. https://doi.org/10.1016/j.bmc.2012.07.046
- Zhang, T., Tong, X., et al. Front. Pharmacol. 2021, 12, 731453. https://doi.org/10.3389/fphar.2021.731453
- https://cresset-group.com/science/overview#xedff


