
基于配体的虚拟筛选发现高亲和力淀粉样蛋白配体
摘要:纤维蛋白聚集是神经退行性疾病的特征,但也是难以进行配体设计的代表性靶标,因为关于结合位点的结构信息有限。在本文中,用基于配体的虚拟筛选开发一种用于筛选Aβ(1-42)纤维新配体的计算方法,并且经实验验证,发现了5个亲和力在nM水平的新配体。首先从文献中收集已知的Aβ(1-42)纤维配体来构建数据库用于训练基于化学结构预测解离常数的模型。然后进行虚拟筛选,虚拟筛选工作流由三个步骤组成:基于电荷、分子量和logP的分子性质过滤器;基于简单化学描述符的机器学习模型;以及使用Forge软件中的场点作为形状和表面性质3D描述符的机器学习模型。这三步工作流用于虚拟筛选ZINC15数据库中的6.98亿个化合物。从亲和力预测值最高的前100个化合物中,使用硫黄素T荧光置换法对其中46个化合物进行了实验研究。鉴定出了5个解离常数在20-600nM范围之间的新配体,它们具有新颖的结构类型,这表明了这种基于配体的方法在发现新的结构独特的高亲和力淀粉样蛋白配体方面的强大功能。使用这种虚拟筛选方法的实验命中率为10.9%。
原文:Timothy S. Chisholm, Mark Mackey, and Christopher A. Hunter. Discovery of High-Affinity Amyloid Ligands Using a Ligand-Based Virtual Screening Pipeline. Journal of the American Chemical Society. 2023,145(29): 15936-15950. DOI: 10.1021/jacs.3c03749
编译:肖高铿
前言
淀粉样蛋白是一类生物分子,它们自组装成具有跨β折叠结构的纤维结构。淀粉样蛋白形成的不溶性蛋白聚集体是许多疾病(尤其是神经退行性疾病)的标志1-3。最常见的神经退行性疾病阿尔茨海默病(AD)的特征是淀粉样斑块的沉积,这些淀粉样斑块由错误折叠的淀粉样β(Aβ)肽和错误折叠的tau蛋白组成的神经原纤维缠结(NFT)组成4,5。虽然40个残基的Aβ肽Aβ(1-40)是脑中最丰富的同工型,但42个残基的Aβ肽Aβ(1-42)是AD中淀粉样斑块的主要成分6。这些肽是通过切割膜蛋白淀粉样前体蛋白(APP)产生的7。然而,APP和Aβ肽的精确生理作用及其在疾病发生和进展中的作用尚不清楚8,9。
目前确定诊断阿尔茨海默病的唯一方法是通过对Aβ斑块进行死后组织病理学鉴定10。由于对活体大脑的获取受限,在临床上使用认知测试以及成像或生物流体测试来诊断阿尔茨海默病11。然而,这些诊断方法只有在大面积的神经元损伤已经发生时才能可靠地诊断阿尔茨海默病10。准确的早期诊断对于确保患者接受适当的疾病管理和在开发疾病疗法时准确招募临床试验人群至关重要12。一个有前景的诊断策略是使用正电子发射断层扫描检测体内的淀粉样蛋白斑块13-17。文献中报道了几种放射性标记的PET探针用于成像淀粉样蛋白斑块18-21。一些PET探针已被批准用于临床,但显示其灵敏度和特异性不足以自行确定诊断阿尔茨海默病17,22,23。淀粉样蛋白配体用于体外实验,包括成像和表征淀粉样蛋白沉积物以及监测蛋白质聚集24,25。 对于这些应用,需要选择性地与靶纤维结合的高亲和力淀粉样蛋白配体。
迄今为止,淀粉样蛋白配体主要通过高通量筛选结合结构-活性关系(SAR)研究而发现。这些方法已经成功识别出了新结构类型的淀粉样蛋白配体,并产生了高亲和力的结合物,但是需要大量的资源和时间。虚拟筛选(Virtual screening,VS)是一种可以提高发现活性配体效率的方法26-28。VS是一种计算技术,旨在通过使用靶标(基于结构的VS)或已知活性分子(基于配体的VS)的知识来识别活性分子29-30。基于结构的VS通常需要高分辨率的靶标结合位点结构。随着低温EM技术的进步,结构信息最近才可用于淀粉样蛋白纤维31,32。然而,淀粉样蛋白配体与纤维的结合不是一个简单的过程:存在多个结合位点,多个配体之间可能发生直接相互作用33-39。在此之前,人们已经从盲对接研究中发现了淀粉样蛋白结合配体40,尽管这种方法的结果并不总是反映实验结果41,42。因此,精确的基于结构VS需要配体结合位点的结构知识,这对于淀粉样蛋白纤维来说仍然是一个未解决的问题。
基于配体的VS不需要了解靶标的结构或结合位点43-46。相反,用已知活性(和非活性)的结构-活性数据来预测结合。以前的研究已经开发了基于配体的药效团来模拟二苯乙烯和黄酮类似物与淀粉样蛋白纤维的结合47-48。由于有大量文献报道了淀粉样蛋白配体的结合数据,这使得基于配体的VS方法寻找新型高亲和力配体具有吸引力。在本文中,我们描述了一个使用基于配体的VS方法的三步流程来识别新型高亲和力淀粉样蛋白配体。该方法成功地鉴定出了5个新的配体,它们对Aβ(1-42)纤维表现出nM级别的结合亲和力。
方法
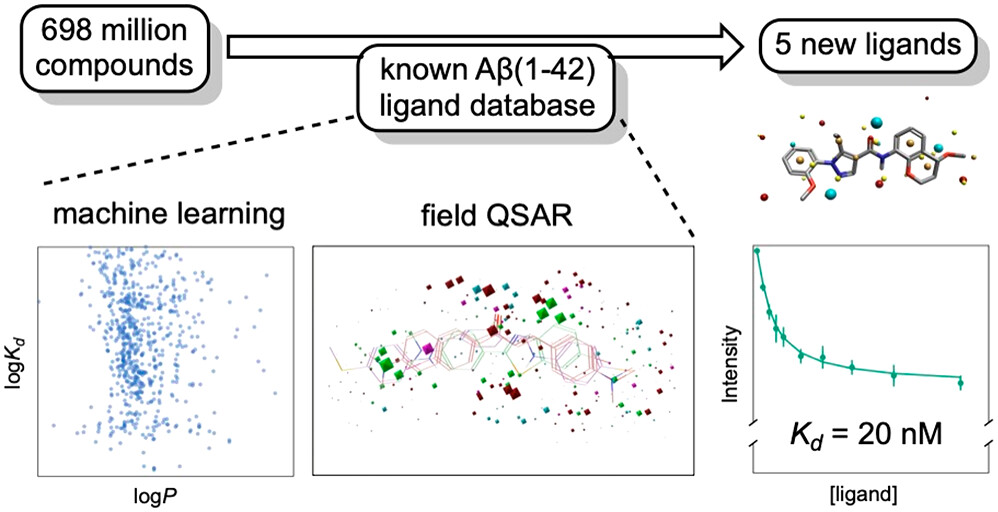
图1展示了使用基于配体的VS发现新淀粉样蛋白配体的步骤。首先,根据logP(辛醇/水分配系数)、分子量和电荷对潜在配体数据库进行过滤。然后,使用机器学习开发一个模型,该模型使用多个化学描述符描述已知配体的离解常数。最后,训练一个包含配体3D分子场信息的更复杂的模型,以预测已知配体的离解常数。在这篇论文中,我们描述了基于已知Aβ(1-42)配体数据集的VS模型的开发,以及这些模型在筛选ZINC15数据库49中的6.98亿个化合物中的应用。将基于配体VS确定的最有前景的、结构多样先导化合物子集进一步进行实验分析以确认与Aβ(1-42)纤维结合,发现了许多新的淀粉样蛋白配体(苗头化合物)。

图1. 基于配体VS的三步工作流。首先,用对ZINC15数据库使用性质过滤器以选择出与已知淀粉样蛋白配体相似的化合物子集。接着用FBH数据库中已知淀粉样蛋白配体的结合亲和力开发的两个模型,并对该子集进行筛选。然后,对从VS模型中获得的亲和力预测值最高的配体进行实验筛选,以确定苗头化合物。
结果与讨论
配体数据库
我们首先汇编了实验确定的配体与Aβ(1-42)纤维结合的离解常数数据集50-183。这个初始数据集总共包含了707个不重复的配体,它们结构多样,并表现出μM至nM的离解常数(Kd)(表S3,表S4)。在这些配体中,44个的Kd值报告为极限值(例如,Kd大于1μM)。图2a显示了剩余663个配体测量的离解常数分布。在这些配体中,许多已被报道靶向Aβ(1-42)纤维上的不同结合位点33-38,50,为了构建基于配体VS的准确预测模型,只应使用结合于同一同结合位点的配体。
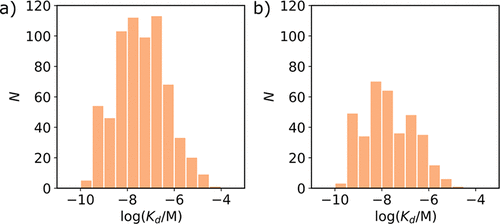 图2.频次分布显示了与(a) Aβ(1-42)纤维和(b) Aβ(1-42)纤维FBH位点结合的实验Kd值的配体数量(N)。如果单个配体报告了多个Kd值,则使用平均值。
图2.频次分布显示了与(a) Aβ(1-42)纤维和(b) Aβ(1-42)纤维FBH位点结合的实验Kd值的配体数量(N)。如果单个配体报告了多个Kd值,则使用平均值。
识别与同一位点结合的配体的最佳方法是竞争性分析实验,用于研究与Aβ(1-42)纤维结合的最常见竞争性分析实验涉及放射性标记的6,5-苯并杂环(6,5-benzoheterocycle,FBH)的置换(见图3中的配体1-4)。直接结合分析中配体1-4测量的离解常数与使用这四种配体中的任何一对进行竞争性分析测量的离解常数相同51-56。这一结果表明,配体1-4全部结合在纤维的同一位点,我们将其称为FBH位点。同样,直接结合分析中配体5-8测量的离解常数与使用配体1-4中的任何一对进行竞争分析测量的离解常数相同67-71。因此,我们得出结论,在竞争分析中置换任何放射性标记的配体1-8测量的离解常数必须报告为与FBH位点的结合。
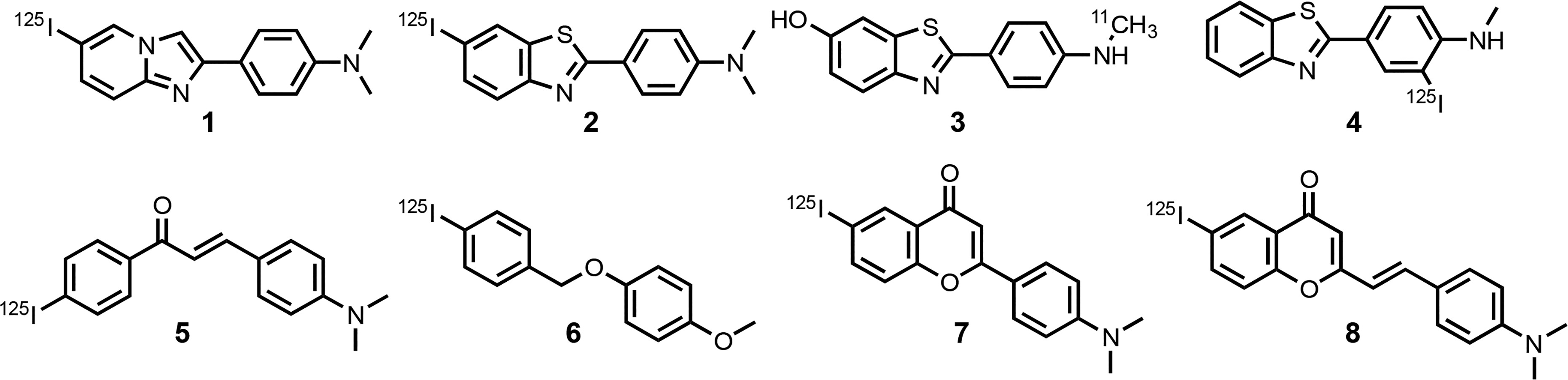
图3.竞争性分析实验的放射性标记配体,用于报告Aβ(1-42)纤维上的FBH结合位点。
先前的工作表明,Aβ(1-42)纤维上存在高亲和力和低亲和力的FBH结合位点,但涉及1-8置换的竞争性实验主要报告为高亲和力位点53,55,124。我们在Aβ(1-42)数据集中共识别出388个配体,这些配体使用与1-8之一的竞争性实验进行表征(表S3)。在这些配体中,27个结合常数报告为极限值。其余361个配体表现出与完整663个配体数据集相似的离解常数范围(比较图2a和2b),并且它们具有多样的化学结构(见图4)。因此,388个FBH位点配体的数据库构成了图1所示的基于配体VS工作流的良好起点。
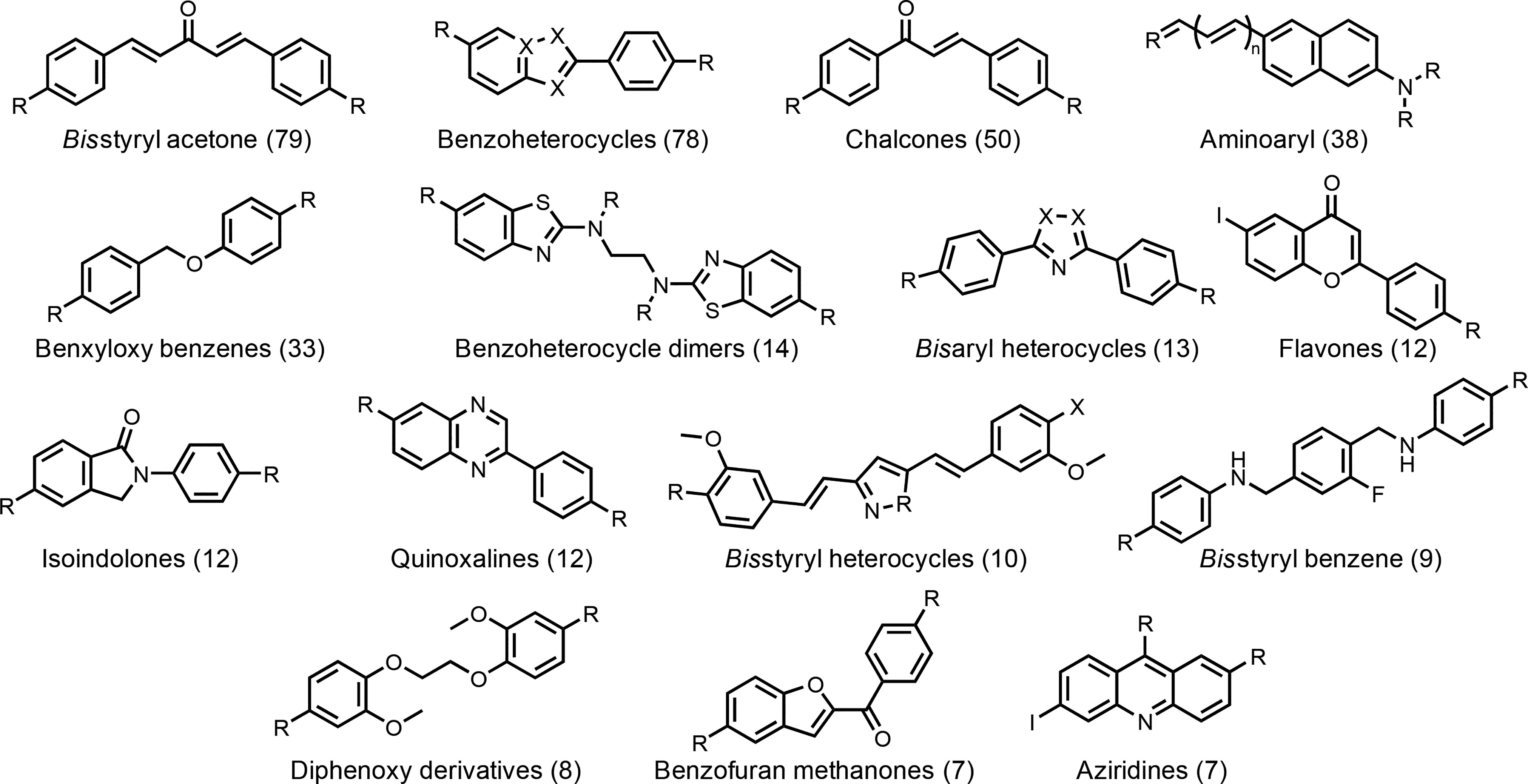
图4. 与Aβ(1-42)纤维FBH位点结合的常见配体结构类型。括号中的数字显示了每种结构类别报告的不重复化合物数量。R和X代表结构变化的位点。
分子性质过滤
图1流程的第一步是根据分子性质过滤ZINC15数据库中的化合物。FBH数据库中只有两个配体含有可电离官能团,因此排除了所有带电荷的化合物。FBH数据库中配体的分子量在200-500 Da范围内,因此过滤掉分子量不在此范围内的任何化合物(图5a)。图5b显示,虽然结合亲和力和logP之间没有相关性,但logP似乎有一个临界值,在数据库中低于该临界值的配体很少。因此,进行第三次过滤以排除logP在3.5-5.5范围之外的化合物,将筛选的化合物总数从6.98亿减少到6300万。
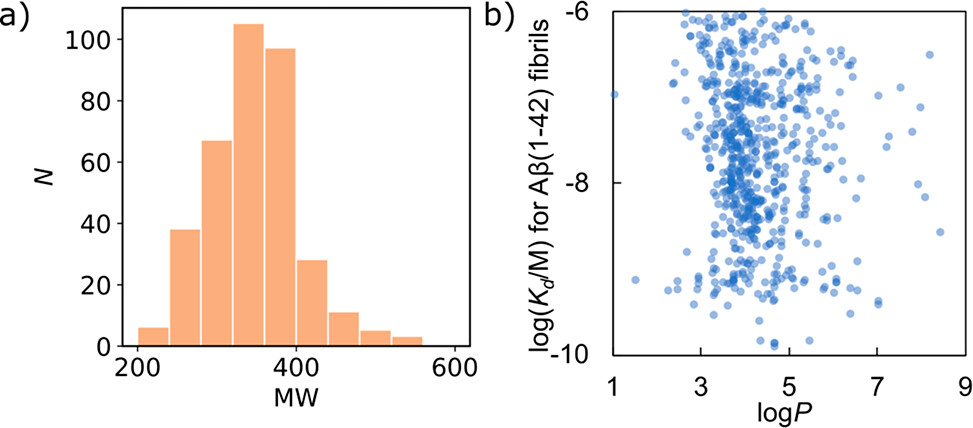
图5. FBH数据库中配体的分子性质。(a)分子量的频率分布。(b)Kd与logP值之间的关系。
分子描述符模型
图1流程的第二步是基于FBH数据库中配体的化学描述符构建模型。使用Python软件包RDKit184为每个配体计算了总共45个不同的描述符,这些描述符基于1D性质(如分子量)、2D拓扑性质(如拓扑极性表面积)和3D构象性质(如非球面度)。为了获得3D描述符,使用Forge中的XedeX构象搜寻算法185为每个配体计算了三维结构。描述符的所有成对比较Pearson相关系数均小于0.9,这表明它们可以被视为独立变量。每组描述符X都使用方程1进行了标准化,以使单位标准差分布以零为中心。
$$X’={X-μ\over σ}···(1) $$
其中μ和σ是数据集中描述符X的平均值和标准差。
然后使用SciKit-learn实现的不同机器学习方法来开发FBH数据库中log(Kd/M)值的预测模型186。基于决策树和增强树(boosted tree)以及支持向量机的机器学习模型在嵌套交叉验证(CV)过程中使用k折验证(内外循环均为k=5,参见SI)187。通过计算log(Kd/M)的预测值和实验值之间的平均误差(MAE)来对回归模型进行打分。然而,有27个解离常数为极限值,不能轻易地纳入回归模型,因此通过将每个配体的log(Kd/M)转化为结合类别来开发分类模型:log(Kd/M)小于等于-8为0类,在(-8,-7]为1类,在(-7,-6]为2类,大于-6为3类。分类模型通过计算平衡准确度(balanced accuracy)进行打分,不同模型的打分结果如表1所示。
表1. 对FBH数据库中配体log(Kd/M)预测模型的评估a
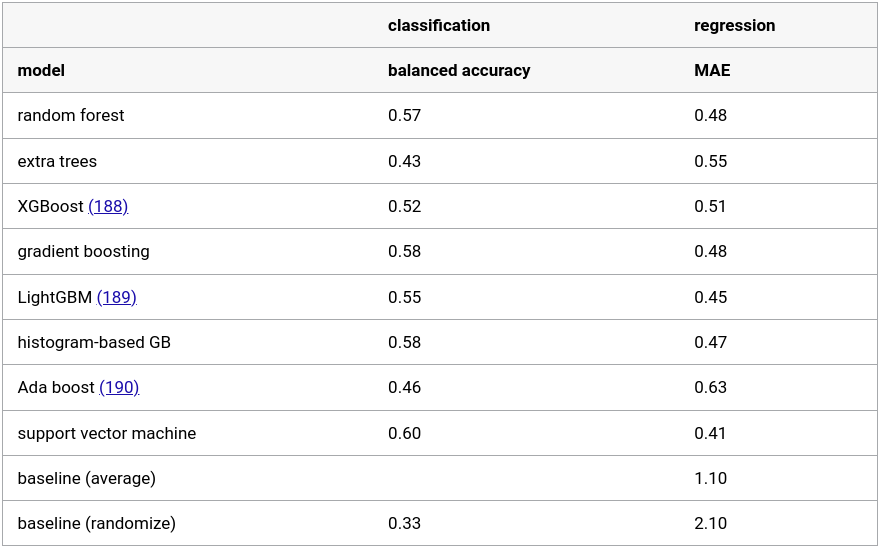
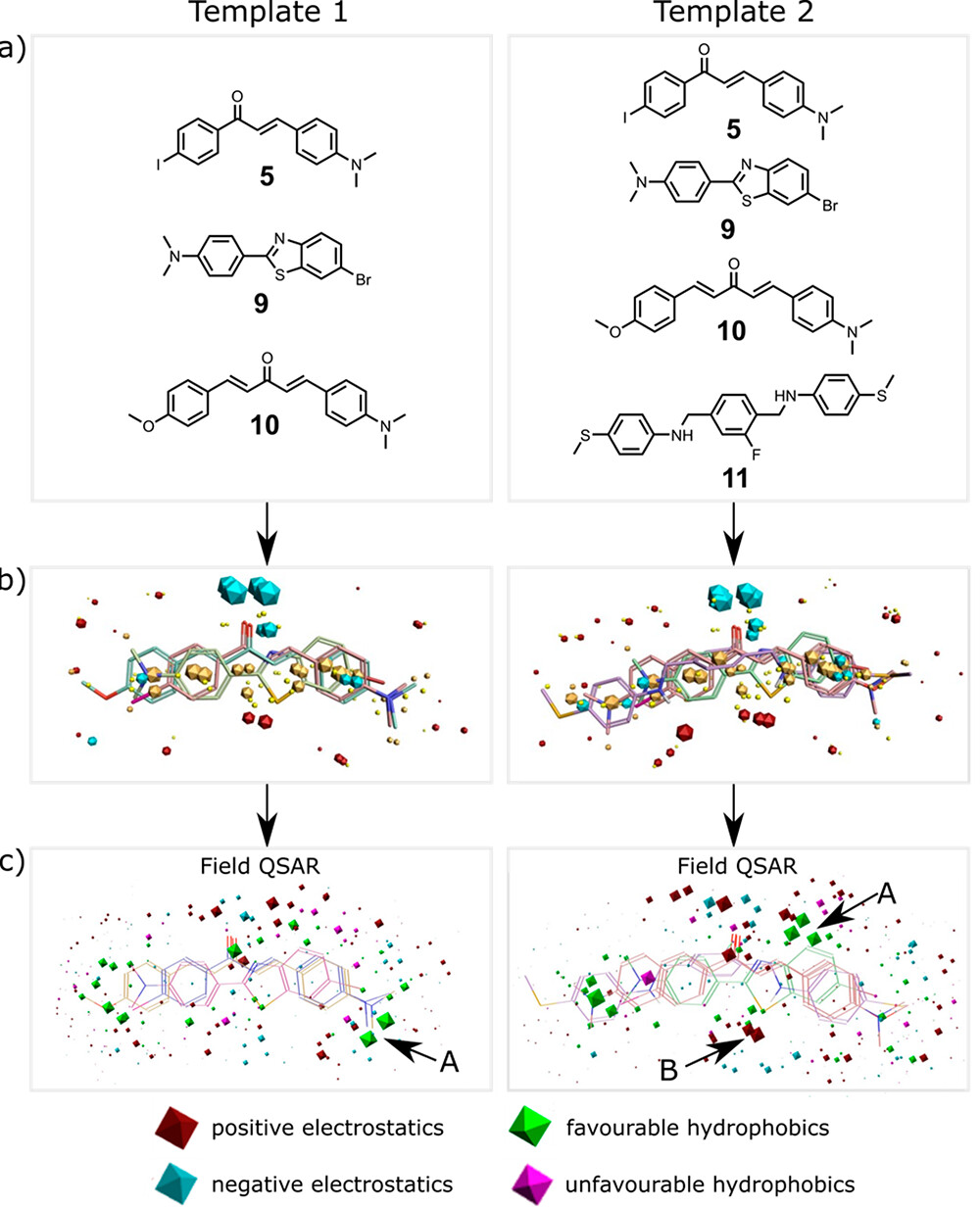
图6. 场点模板。(a)参比配体5、9和10用于构建模板1,参比配体5、9、10和11用于构建模板2。(b) 参比配体的3D叠合及其场点(蓝色:负静电势;红色:正静电势;橙色:高疏水性;黄色:范德华相互作用)。(c) Field-QSAR模型(相对于参比配体):蓝色区域描述了负静电场系数越负或正静电场系数越小对亲和力有利的区域;红色区域描述了负静电场系数越小或正静电场系数较大对亲和力有利的区域;绿色区域描述了疏水性有利于亲和力的区域;粉红色区域描述了疏水性不利于亲和力的区域。标记为A的区域表示疏水相互作用重要的区域,标记为B的区域表示静电相互作用重要的区域。
图6a中所示的配体5、9、10和11用于构建模板,因为它们具有纳摩尔级的结合亲和力和在FBH数据库中频繁出现的结构母核。可以将所有四个配体叠合创建模板,但这需要有高能构象(图6中的模板2)。如果排除配体11,则可以以低能构象叠合其他三个配体(图6中的模板1)。然后,将来自FBH数据库结构相似的配体叠合到这些模板上:对于模板1,添加苯并杂环、氨基芳基、黄酮、喹喔啉、苄氧基苯、查耳酮、双芳基杂环和双苯乙烯基丙酮;对于模板2,添加双苯乙烯基杂环和双苯乙烯基苯(见图4)。根据化合物根据其叠合到模板之后的形状和场点相似性进行打分。人工检查叠合以确保每个结构类别的一致性。某些官能团证明难以叠合,例如,含有乙二醇链或聚乙烯连接臂的配体产生了大量得分相似但场点分布不同的叠合。这些化合物在模型开发时予以放弃。在对叠合过的数据集进行优化之后,共有212个配体叠合到模板1,222个配体叠合到模板2。
对于每个模板,配体划分为训练集(80%)和测试集(20%)用于模型开发。划分区是按活性分层的,并且是手动执行的,以确保图4中显示的每个配体结构类别在训练集和测试集中都有代表性化合物。使用Forge为每个模板构建了五个不同的QSAR模型:随机森林(random forest)、支持向量机(support vector machine)、关联向量机(relevance vector machine)、k-最近邻(k-nearest neighbors,KNN)和基于场点偏最小二乘分析的回归方法(Filed QSAR)192。随机森林、支持向量机和关联向量机模型使用了k-折交叉验证过程(k=5),k-最近邻和场QSAR模型使用了留一法交叉验证。使用训练集和测试集的回归系数r2以及训练集的交叉验证回归系数q2来评估模型性能,结果见表2。
表2. Forge模型的回归系数(r2)和交叉验证回归系数(q2)
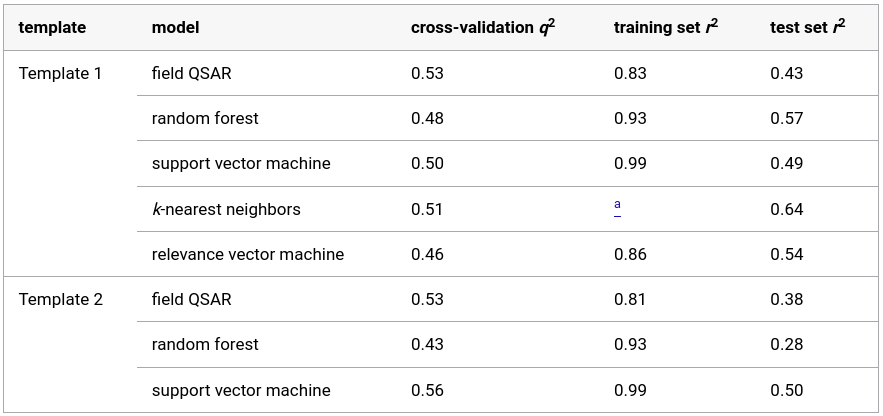
a. 没有应用
不同模型的训练集和交叉验证集的性能表现非常相似。对于测试集,随机森林模型在模板1上,支持向量机模型在模板2上,分别获得最高的r2值。这些模型与Field QSAR模型一起用于筛选使用化学描述符从ZINC15数据库中选出的10,000个化合物。虽然Field QSAR模型的测试集r2值较低,但该类模型非常有用,因为它们识别出了对配体结合亲和力起到决定作用的重要相互相互作用。图6c显示了两个Field QSAR模型。两个模板在疏水相互作用(箭头A)和静电相互作用(箭头B)决定的最重要位点显示出显著差异。
首先使用表示化合物与图6所述模型场点相似程度的Filed QSAR distance-to-model打分(模型距离打分)对10,000个化合物进行过滤。只有模型距离打分良好或优秀的化合物才会被进一步考虑。然后对两个模型(即模板1的随机森林和Filed QSAR以及模板2的支持向量机和Field QSAR)的log(Kd/M)预测值取均值,用于对每个模板的化合物进行单独排名。每个模板log(Kd/M)预测值均值最低的50个化合物被选中,基于化合物的商品可供货情况以及骨架多样性(见SI)从100个化合物中采购了其中46个用于实验筛选。虽然之前文献中报道的Aβ(1-42)配体(图4)通常是刚性且平面性的,但本次虚拟筛选的许多苗头化合物包含有柔性脂肪链和环。
结合实验
硫黄素T(Thioflavin,ThT)竞争性分析用于筛选从VS工作流中选出的46个化合物(图7a)。当ThT与Aβ(1-42)纤维结合时,荧光发射强度大大增强,发射波长发生偏移。在ThT结合位点处添加第二个与Aβ(1-42)纤维结合的非荧光配体L,由于ThT的位移导致荧光强度会降低。因此,将第二个竞争配体(L)滴定到ThT与Aβ(1-42)纤维的混合物中可以确定结合亲和力。图7b显示了这种竞争性分析的一个例子。当ThT在实验的第一阶段与Aβ(1-42)纤维结合时,荧光强度特征性地增加。在实验的第二阶段添加竞争性配体导致荧光强度降低,这是由于大约一半的ThT从结合位点中被置换出来造成的(图7b)。
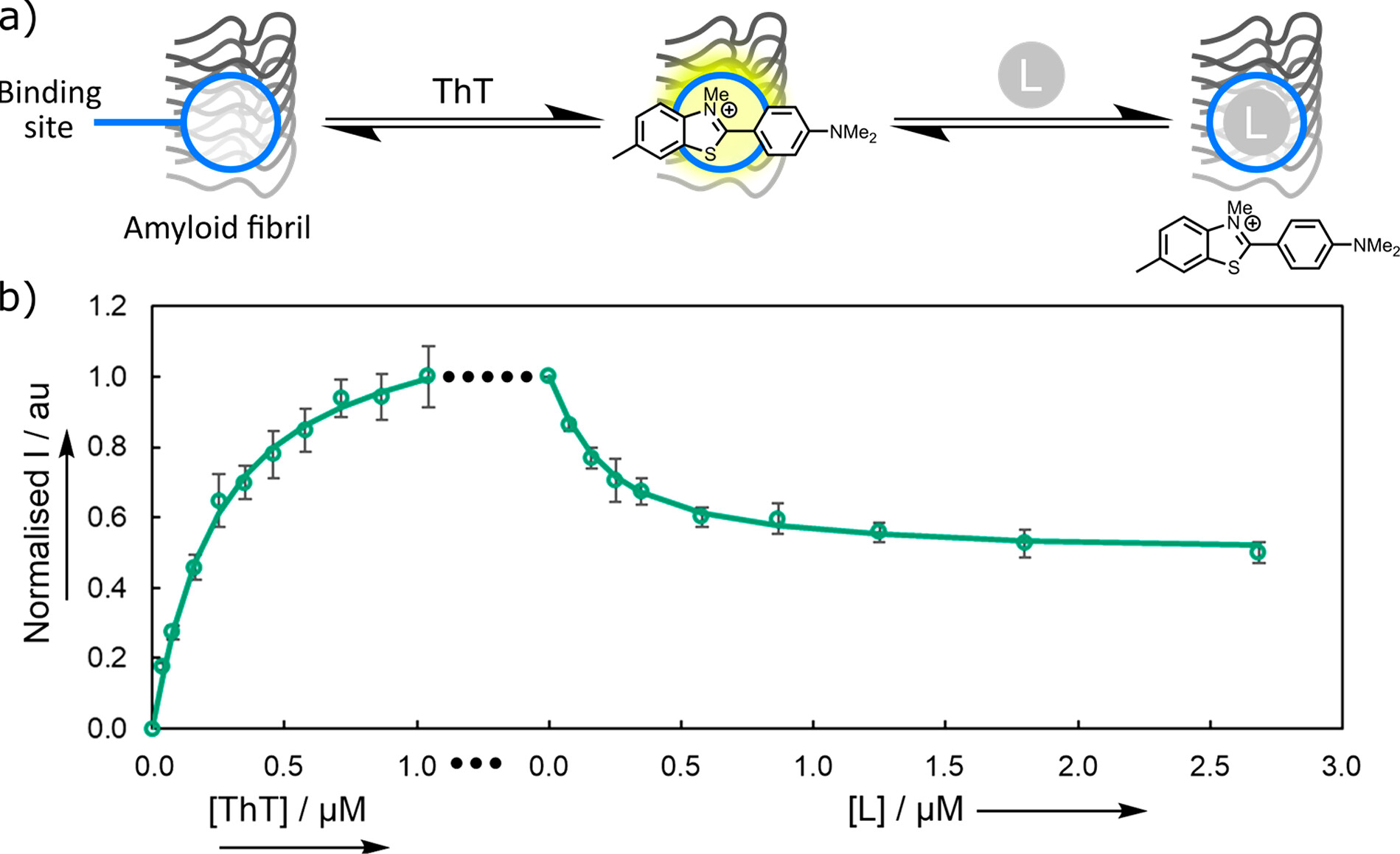
图7. (a)ThT竞争性结合分析。当ThT与淀粉样蛋白纤维结合时,荧光强度增加。当ThT被置换时,通过荧光强度的降低检测到竞争配体L (E570)的结合。(b)将ThT滴入1× PBS缓冲液(pH 7.4,25°C)中的Aβ(1-42)纤维溶液(500 nM),然后滴定竞争性配体。使用λex = 440 nm记录光谱,并在λem = 483 nm下监测荧光发射。实验测量值以点表示,其中误差条表示至少三次独立实验计算的95%置信区间,两条线是与方程2的最佳拟合,其log(Kd(ThT)/M) = -6.7和log(Kd(L)/M) = -7.6。
游离态和结合态的ThT都会发出荧光,因此,在滴定数据分析中必须考虑游离态ThT的背景荧光。此外,必须考虑两种不同类型的结合位点的存在:S1位点可结合ThT和L,S2位点只能与ThT结合。因此,荧光发射的强度(I)由方程2给出:
$$?=ϵ_fΦ_f[ThT]+ϵ_bΦ_b([ThT·S_1]+[ThT·S_2])···(2) $$
其中,εfϕf和εbϕb分别是游离态和结合态ThT的紫外-可见光吸收消光系数和荧光量子产率的乘积,[ThT]是游离态ThT的浓度,而[ThT·S1]和[ThT·S2]分别是结合到S1和S2的ThT浓度。
与每个位点结合的ThT浓度由方程3给出:
$$[ThT·S_?]=?_d(ThT)[ThT][S_?]···(3)$$
其中[Sn]是未结合位点Sn(n=1或2)的浓度,并且假设ThT的离解常数Kd(ThT)对于两个位点都是相同的。
与位点S1结合的L的浓度由方程4给出:
$$[L·S_1]=?_d[L][S_1]···(4)$$
其中[L]是游离态L的浓度,[L·S1]是结合到S1的L的浓度,Kd是解离常数。
ThT、[ThT]tot和L、[L]tot的总浓度则由方程5和6给出:
$$[ThT]_{tot}=[ThT]+[ThT·S_1]+[ThT·S_2]···(5)$$
$$[L]_{tot}=[L]+[L·S_1]···(6)$$
通过在1× PBS缓冲液(pH 7.4,25°C)中的稀释实验测量ThT的 ϵfϕf量,并通过将方程2、3和5拟合到图7b所示实验第一阶段的单位点结合模型来计算ϵbϕb和Kd(ThT)的值,即ThT直接滴定到Aβ(1-42)纤维中(−log(Kd/M)= 6.7±0.1;详细信息请参见SI)。
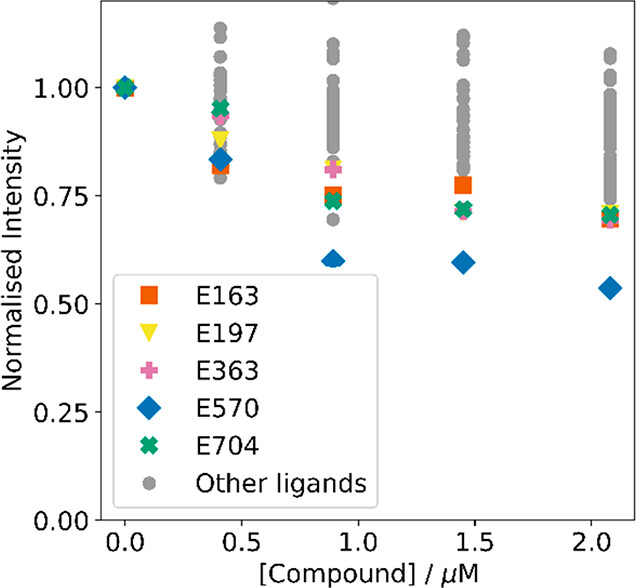
图8. VS工作流中44个化合物的ThT竞争性分析测试。将每个化合物越来越浓地添加到Aβ(1-42)纤维(250 nM)和 ThT(1.0 μM)的1× PBS缓冲液(pH 7.4,25°C)中。使用 λex = 440 nm 记录荧光光谱,并在 λem = 483 nm 下监测发射强度。灰色数据点表示未进一步研究的低亲和力化合物。
图8显示了来自46个VS候选物中的44个滴定到Aβ(1-42)纤维与ThT混合物中的结果。取代ThT最大量的五个化合物(E163、E197、E363、E570和E704)在图8中被高亮显示为彩色数据点,这些化合物将被进一步表征。来自VS的其他两个候选物是荧光香豆素衍生物,因此直接滴定到Aβ(1-42)纤维中用于测定这些化合物,而不是竞争性实验测定。在这些化合物中没有检测到结合(见SI)。
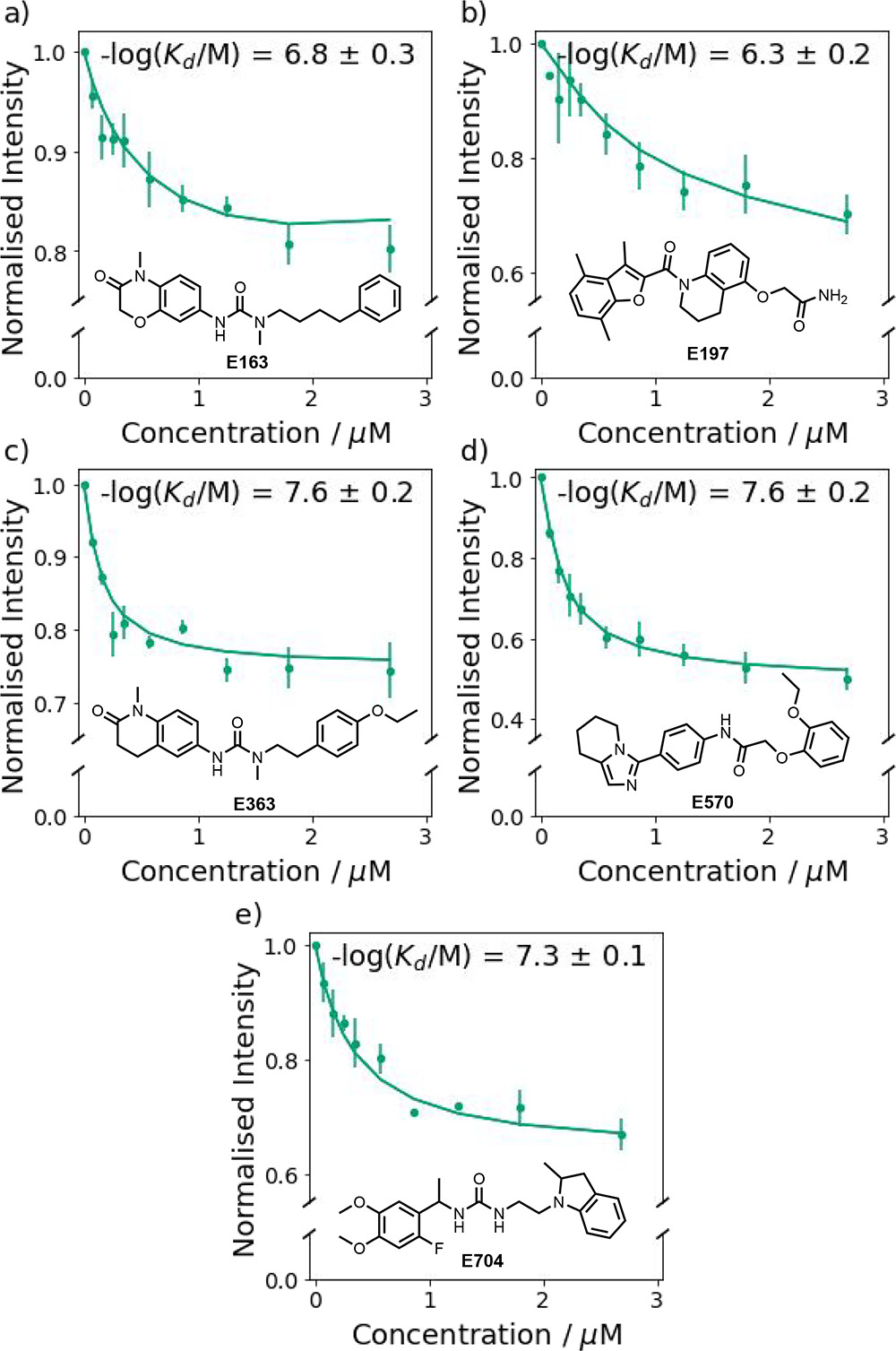
图9. 在1× PBS缓冲液(pH 7.4,25°C)中,将(a)E163,(b)E197,(c)E363,(d)E570和(e)E704滴入Aβ(1-42)纤维(500 nM)与ThT(1.0 μM)的混合物中。使用λex = 440 nm记录光谱,并在λem = 483 nm监测发射。实验测量值以点表示(误差条表示至少三次独立实验计算的95%置信区间)。线条与方程2最佳拟合,并显示了由此得出的离解常数。
图9显示了用于测量E163、E197、E363、E570和E704与Aβ(1-42)纤维的结合亲和力的滴定实验结果。拟合1:1结合等温线得到5个化合物的纳摩尔解离常数(表3,20-600 nM)。ThT置换量因化合物而异,这表明化合物靶向ThT结合位点的不同子集。E570置换约一半的ThT,而其他四个仅置换20-40%的结合ThT。这一结果的一个解释是,用于模型开发的配体可能结合到纤维上的多个位点,因此VS选择的化合物可能包含有利于在不同位点结合的不同特征组合。部分配体置换是这些分析的一个潜在有用特征,可以提供不同类型纤维上存在的不同结合位点的性质和分布的额外信息34。
表3.在Aβ(1-42)纤维(500 nM)和ThT(1.0 μM)混合物的1×PBS缓冲液(pH 7.4,25°C)中加入E163、E197、E363、E570和E704,通过荧光竞争性试验测得的离解常数a
| Compound | Kd | –log(Kd/M) |
|---|---|---|
| E163 | 200 ± 100 | 6.8 ± 0.3 |
| E197 | 600 ± 300 | 6.3 ± 0.2 |
| E363 | 20 ± 10 | 7.6 ± 0.2 |
| E570 | 56 ± 6 | 7.6 ± 0.1 |
a.使用λex = 440 nm记录光谱,并在λem = 483 nm监测发射。至少三个独立实验的拟合平均值给出离解常数。
图10显示了与模板2叠合配体的场点。场点分布之间没有明显的相似性,这与不同的结合位点偏好是一致的,突出了3D模型在寻找结构多样配体方面的实用性。
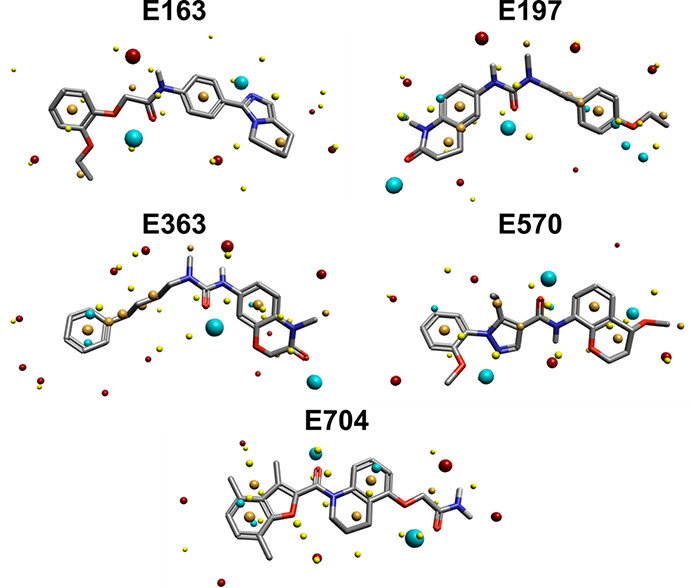
图10. 叠合到模板2配体E163、E197、E363、E570与E704的场点。蓝色场点描述负静电势区域;红色场点描述正静电势区域;橙色场点描述高疏水性区域;黄色场点描述范德华相互作用。
用RDKit指纹计算五个配体与FBH数据库中每个化合物之间的Tanimoto相似系数193,194,图11显示了结果。在所有情况下,相似系数的最大值约为0.5,表明新发现的配体的化学结构与所有先前报道的Aβ(1-42)配体的化学结构有很大不同。图12将E163、E197、E363、E570和E704的结构与FBH数据库中具有最高Tanimoto系数的相应配体进行了比较。
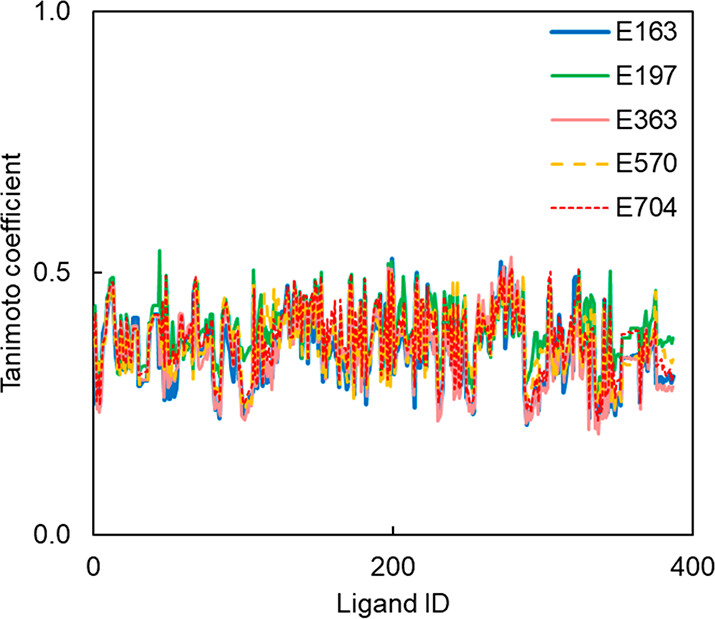
图11. 使用RDKit指纹计算E163、E197、E363、E570、E704与FBH数据库中每个配体之间的Tanimoto相似系数。
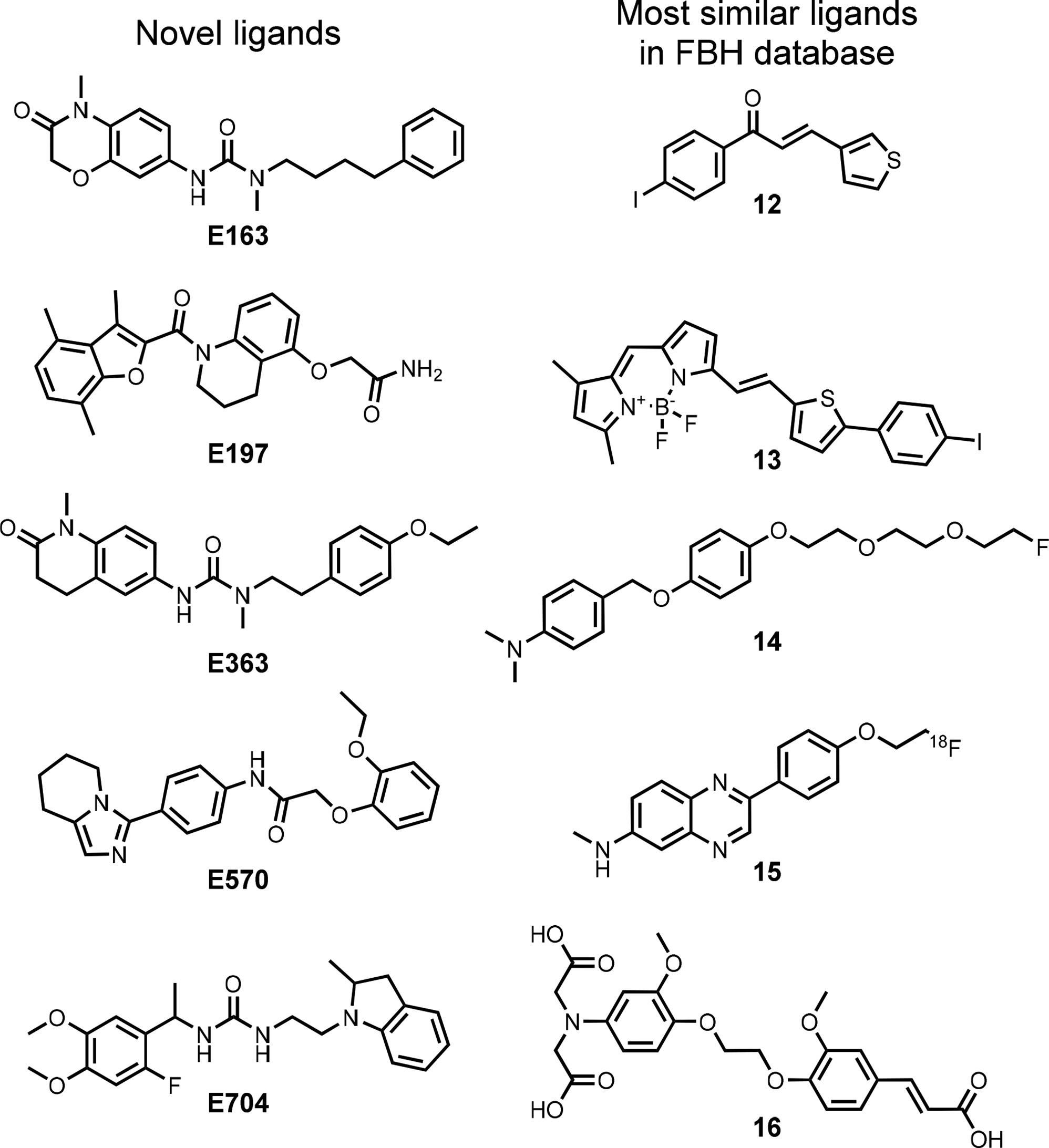
图12. VS工作流识别出的新型配体的化学结构与FBH数据库中相应配体的化学结构进行比较,其最高Tanimoto相似系数为:E163和12为0.53,E197和13为0.54,E363和14为0.53,E570和15为0.50,E704和16为0.51。
结论
由于缺乏有关结合位点位置与结构的知识,淀粉样蛋白纤维(Amyloid fibrils)成为基于结构虚拟筛选(VS)的一个挑战性靶标。在本文中,我们描述了一种三步基于配体的虚拟筛选方法,该方法利用了文献中丰富的Aβ(1–42)配体数据。首先,我们标注了一个包含707个Aβ(1–42)纤维结合配体的数据集,其中388个配体在相同结合位点上有文献报道地结合常数,这些数据是通过配体竞争性实验确定的。从FBH数据库中识别出结合所需的关键分子属性。ZINC15数据库中的6.98亿化合物通过电荷、分子量和logP进行了筛选,从而选出了6300万化合物进行进一步筛选。FBH数据库被用来训练支持向量机,通过使用计算成本低廉的化学描述符来预测解离常数。该模型被用来选择预测亲和力最高的10,000个化合物。FBH数据库被用来训练基于场点的3D模型,这些场点代表了表面、形状和电子属性的描述。
用这些模型选出了结合亲和力预测值最高的100个化合物,其中46个在Aβ(1-42)纤维的荧光竞争结合试验中进行了实验研究。五个亲和力最高的配体在未经进一步结构优化的情况下具有纳摩尔级的解离常数(25-500 nM)。通过基于配体的虚拟筛选工作流筛选出的46个化合物进行实验研究,发现了5个新的淀粉样蛋白配体,其发现命中率为10.9%。虚拟筛选工作流还产生了结构多样的化合物,这些化合物是未见报道的Aβ(1-42)配体新骨架195。新配体的构象柔性也表明,先前报道的Aβ(1-42)配体的刚性、高度共轭结构不是严格必需的。该方法不限于Aβ(1-42)配体。例如,将这种方法应用于配体结合的体内数据,对于加速发现与疾病相关的生物纤维的新型高亲和力配体特别有意义。蛋白质聚集体的新配体在疾病诊断中有许多潜在的应用,包括用作体内成像剂或鉴定组织样本中的不同纤维多晶型。
支持信息(Supporting Information,SI)
支持信息可免费获得:https://pubs.acs.org/doi/10.1021/jacs.3c03749
参考文献
忽略,请参见原文:https://pubs.acs.org/doi/10.1021/jacs.3c03749。
联系我们,获取软件试用或商务合作
本文所用的软件Forge现在是Flare一部分,您可以联系我们获取试用版,在自己的项目中亲自尝试同样的机器学习与Filed QSAR技术。
原创文章,作者:小墨,如若转载,请注明出处:《基于配体的虚拟筛选发现高亲和力淀粉样蛋白配体》http://blog.molcalx.com.cn/2024/03/09/amyloid-ligand.html


