
摘要:本文展示了Flare FEP作为一种可靠且用户友好的配体结合自由能预测方法。Furui和Ohue证明了百度的HF3模型在预测apo蛋白结构(无配体)和holo蛋白-配体复合物(有配体)方面的有效性。通过将HF3与Flare FEP结合,他们成功实现了结合自由能的无缝预测,突显了Flare FEP在方法验证中的潜力,并能够将其无缝整合到计算机辅助药物设计工作流程中。
作者:Matthew Kondal/March 17, 2025
编译:肖高铿
前言
精确的蛋白质-配体结合自由能计算是计算机辅助药物发现中的重要工具,因为它们能够识别和优选高活性化合物,从而降低实验成本。自由能微扰(FEP)是计算结合自由能的黄金标准方法,但其准确性在很大程度上取决于输入的蛋白质-配体复合物结构的质量,同时它还会带来较高的计算成本以及潜在的高额经济成本。
AI驱动的蛋白质结构预测模型提供了一种利用现有蛋白质结构数据生成同源模型的方法,这些模型可用于计算尚无晶体结构体系的配体结合自由能。然而,这种方法仍然高度依赖于同源模型的准确性。尽管像AlphaFold2这样的模型在重现与晶体结构相当的结合自由能方面取得了一定的成功,但其无法预测蛋白质复合物(holo)结构的局限性,限制了其在高效FEP工作流程中的直接应用1。
最新的AlphaFold3在与小分子配体、核酸、离子以及修饰残基的复合物结合结构预测方面展现了最先进的性能2。当AlphaFold3仅作为网络服务可用时,百度的团队3开发了蛋白质结构预测模型HelixFold3(HF3)。HF3旨在模拟AlphaFold3,同时提供了一个更易于获取的蛋白质-配体复合物预测解决方案,并保持了较高的准确性。随后,Furui和Ohue等人使用Flare FEP验证了其模型的实用性4。 在本文中,我们将展示Flare的FEP解决方案,以及如何高效地将其应用于由HF3生成的蛋白质模型的配体结合自由能预测,从而验证他们的蛋白质-配体结构预测方法。
用Flare FEP计算结合自由能
FEP(自由能微扰)是一种用于预测分子结合自由能的计算技术。准确且高效地计算这些值使得研究人员能够在药物发现过程的早期优先选择最有潜力的候选分子,从而减少与实验测试相关的时间和成本。Cresset处于数字化化学解决方案的前沿,通过Flare FEP提供先进的相对和绝对FEP方法。
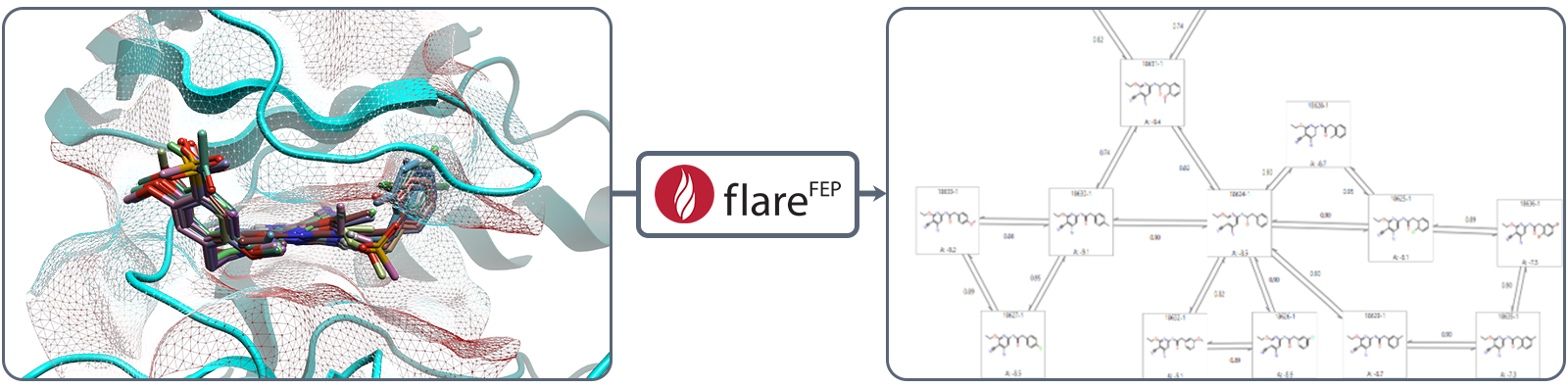
图1. JNK1衍生物及其结合位点,显示了蛋白静电势表面以及用Flare FEP自动生成的配体FEP图,其中配体数据来源于Furui等人论文的支持材料。
相对结合自由能(RBFE)计算是FEP的一种形式,它计算化学相关配体之间的结合自由能相对于彼此的差异。其工作原理是首先生成一个FEP图(图1),将化学结构相似的配体连接起来,然后通过炼金术路径将一种配体转化为另一种,从而计算出它们之间的自由能变化。这一过程非常复杂,但Flare FEP的设计目标是使这些步骤尽可能准确、高效且易于使用。因此,已通过Cresset大幅改进的LOMAP5版本实现自动化的图生成,并且如果图中存在过于复杂的转化,无法在单一步骤内完成,Flare能够智能识别并插入中间体以连接分子。
用于在两个终态之间进行转化的炼金术路径由一系列lambda窗口组成,由于每个lambda窗口包含一个短时分子动力学模拟,所使用的lambda窗口数量会极大地影响计算效率。Flare FEP使用“自适应lambda窗口算法”6,自动评估并应用每个转化所需的最优lambda窗口数量,以实现FEP计算的收敛,通常可减少30%的模拟窗口数量。这些特性确保Flare FEP保持准确性、可靠性、高效性以及用户友好性,使其非常适合无缝采用和探索性使用。
用Flare FEP验证HelixFold3预测的蛋白质-配体复合物结构
最成功的FEP项目依赖于对靶标体系的深入理解以及蛋白质-配体结构的准确表征。在理想情况下,得有一个高分辨率的蛋白质-配体复合物,以确保自由能计算是基于生物学相关的构象进行的。然而,由于这些信息并不总是可以获得,这时可以使用AI模型来进行预测。
Furui和Ohue使用HF3预测了Wang等人7FEP基准数据集中8个靶标(BACE、CDK2、JNK1、MCL1、P38、PTP1B、凝血酶、TYK2)的结构,并将它们与实验得出的晶体结构进行了比较。首先,他们为每个靶标预测了五个apo(无配体)和holo(有配体)结构,从中选择了一个,并评估了三个RMSD值:配体RMSD、全局RMSD以及结合位点RMSD。他们发现,在所有结构中,配体RMSD均低于2 Å,holo预测结构的全局RMSD普遍低于apo结构(BACE和JNK1除外)。通过比较apo和holo结构的结合位点RMSD,发现CDK2、MCL1、P38和PTP1B的holo结构相较于apo结构有所改善,而其他靶标的RMSD则基本保持不变。尽管JNK1和P38的结合位点RMSD有所改善,但仍然高于2 Å,这表明HF3在准确预测这些结合位点方面存在困难(以RMSD为衡量标准)。
计算结合自由能为预测的蛋白质-配体复合物的准确性提供了更为实用的评估方法。因此,Furui和Ohue使用晶体结构、HF3 apo(无配体)和HF3 holo(有配体)蛋白结构计算了数据集中配体的结合自由能,并将其与实验值进行了比较。他们对Wang等人数据集的分析揭示了模型性能的一些差异。凝血酶(Thrombin)始终表现出最强的预测准确性,其R²范围为0.856至0.882,MUE(平均无符号误差)在0.152至0.381 kcal/mol之间。相比之下,BACE的表现始终最差,其R²较低(0.123至0.325),MUE较高(0.986至1.007 kcal/mol),而晶体结构的PTP1B的单个R²最低,仅为0.002。尽管存在这些波动,如果排除晶体结构的PTP1B和HF3 apo MCL1复合物,基于10,000次自助抽样(bootstrap sampling)计算的重叠90%置信区间表明,HF3与Flare FEP能够以与晶体结构相同的精度预测自由能。
HF3是基于PDB中的结构进行训练的,因此其训练数据中会包含Wang等人数据集中的结构。为了挑战他们的模型,Furui和Ohue预测了数据集中衍生结构的模型,这些衍生结构使用了训练数据中未包含的配体进行建模。结果表明,HF3能够有效地预测大多数衍生物的配体位置,RMSD值通常保持在2 Å以下。然而,JNK1的衍生物表现出显著偏差,21个衍生物中有8个的RMSD值超过了这一阈值。图2展示了JNK1晶体结构与最不匹配配体的叠合,突出了HF3预测偏离的部分。进一步观察表明,这种差异源于晶体结构中的配体缺少一个氯苯基团,而在我们根据支持信息中的项目文件重建的模型中,该氯苯基团更深入地嵌入了蛋白质口袋(图2)。尽管存在这种差异,Flare FEP仍能够合理预测整个衍生物集合中配体的结合自由能,除MCL1的MUE为1.5 kcal/mol外,其他靶标的MUE均小于1 kcal/mol。
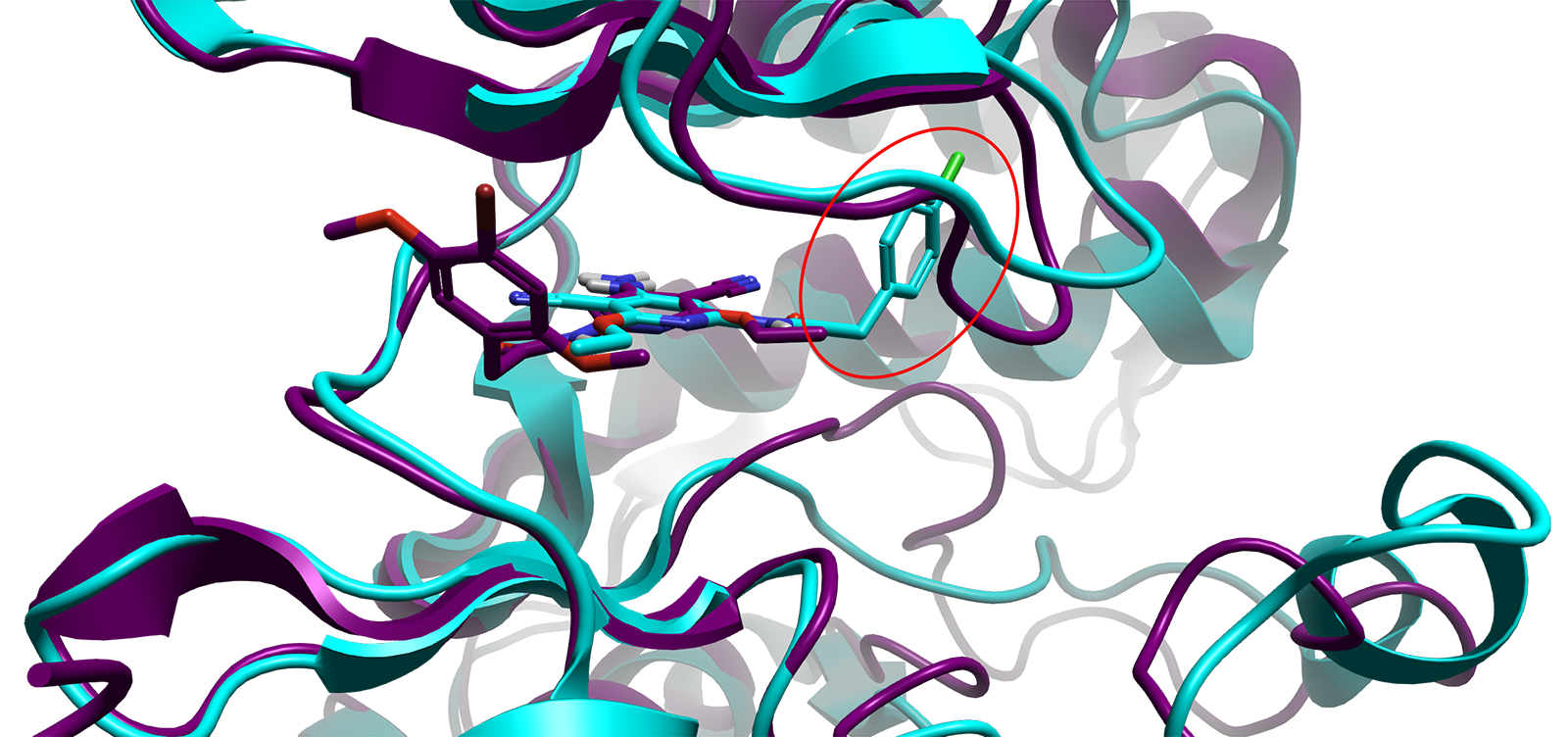
图2. JNK1的叠合结构。紫色部分是HelixFold3预测的结构,配体为18627-1;青色部分是晶体结构,配体为17124-1。该图像使用Furui和Ohue在补充信息中提供的文件创建。
结论
本文展示了Flare FEP作为一种可靠且用户友好的配体结合自由能预测方法。Furui和Ohue证明了百度的HF3模型在预测apo蛋白结构(无配体)和holo蛋白-配体复合物(有配体)方面的有效性。通过将HF3与Flare FEP结合,他们成功实现了结合自由能的无缝预测,突显了Flare FEP在方法验证中的潜力,并能够将其无缝整合到计算机辅助药物设计工作流程中。
参考文献
- Beuming, T.; Martín, H.; Díaz-Rovira, A. M.; Díaz, L.; Guallar, V.; Ray, S. S. Are deep learning structural models sufficiently accurate for free-energy calculations? Application of FEP+ to AlphaFold2-predicted structures. J. Chem. Inf. Model. 2022, 62, 4351–4360. https://doi.org/10.1021/acs.jcim.2c00796
- Abramson, J., Adler, J., Dunger, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 2024, 630, 493–500. https://doi.org/10.1038/s41586-024-07487-w
- Liu, L.; Zhang, S.; Xue, Y.; Ye, X.; Zhu, K.; Li, Y.; Liu, Y.; Zhang, X.; Fang, X. Technical Report of HelixFold3 for Biomolecular Structure Prediction. arXiv preprint, arXiv:2408.16975 , 2024.
- Furui, K., Ohue M., Benchmarking HelixFold3-Predicted Holo Structures for Relative Free Energy Perturbation Calculations. ACS Omega 2025. https://doi.org/10.1021/acsomega.4c11413
- Liu, S., Wu, Y., Lin, T. et al. Lead optimization mapper: automating free energy calculations for lead optimization. J. Comput. Aided Mol. Des., 2013, 27, 9, 755-770. https://doi.org/10.1007/s10822-013-9678-y
- Scott D. Midgley, Sofia Bariami, Matthew Habgood, and Mark Mackey, Adaptive Lambda Scheduling: A Method for Computational Efficiency in Free Energy Perturbation Simulations. J. Chem. Inf. Model. 2025 65 (2), 512-516. https://doi.org/10.1021/acs.jcim.4c01668
- Wang, L. et al. Accurate and Reliable Prediction of Relative Ligand Binding Potency in Prospective Drug Discovery by Way of a Modern Free-Energy Calculation Protocol and Force Field. J. Am. Chem. Soc. 2015, 137, 2695–2703. https://doi.org/10.1021/ja512751q
联系我们
Flare V10是Flare系列软件的最新版本,交付先进的科学方法、分析工具和直观、易用的增强功能,洞察您的配体 – 蛋白质复合物结构。
想要尝试Flare信息丰富、用户友好界面,发现它如何帮助您自信地推动潜在先导化合物优化?请现在就联系我们安排试用,快速访问Flare的广泛功能。我们的专业团队随时准备通过安装和设置为您提供支持,而我们全面的教程库——涵盖从常见工作流程到高级方法和功能的所有内容将帮助您开始使用。我们在这里帮助您更快地实现目标,让您设计出感兴趣的分子。
电邮:info@molcalx.com
电话:020 – 38261356


