
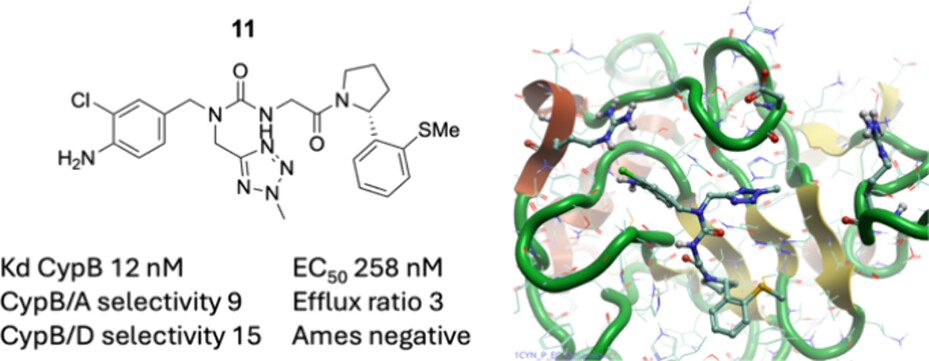
摘要:代谢功能障碍相关脂肪肝病(MASLD)是全球范围内日益严重的公共卫生问题,其进展可导致肝硬化、肝衰竭及肝癌。尽管2024年FDA批准了首个MASH药物雷米特罗,但针对纤维化相关靶点的治疗仍存在挑战。本研究聚焦于亲环素B亚型(Cyp B)抑制剂的开发,因其在线粒体凋亡和胶原生成中的关键作用,Cyp B被认为是抗纤维化治疗的重要靶点。通过基于结构的计算模型(Flare FEP),在三臂亲环素抑制剂1为起点,重点针对Abu、Pro和3点钟方向口袋进行各种取代基替换的FEP扫描,并将FEP计算结合亲合力高的化合物进行组合、FEP计算预测靶标选择性等评估。最后经过合成与测试,化合物11脱颖而出,展现出对Cyp B的强效抑制(Kd≈10 nM)及高达80倍的Cyp B/A选择性。机制解析揭示,其选择性源于Cyp B特异性残基(如Arg90、Asp79)与配体的互补相互作用。细胞实验表明,化合物11显著降低了iPSC衍生肝细胞的脂质沉积(EC50≈0.3 μM),效果与FDA批准药物雷米特罗相当,且未观察到明显细胞毒性。DMPK分析显示,化合物11具有良好药代动力学特性(血浆蛋白结合率低、CYP酶抑制风险可控)及无致突变性,安全性优势突出。本研究首次证实,选择性Cyp B抑制剂可通过独立于THRβ的机制干预MASH脂质代谢,为联合疗法提供了新策略。化合物11的优化过程凸显了Flare FEP计算驱动药物设计在解决亲环素抑制剂选择性难题中的潜力,为后续临床开发奠定了基础。
原文:Kouridaki, M.-E. et al. (2025) “Optimization of Cyclophilin B-Targeted Tri-vector Inhibitors for Novel MASH Treatments,” Journal of Medicinal Chemistry. Available at: https://doi.org/10.1021/acs.jmedchem.5c00301.
编译:肖高铿
前言
代谢功能障碍相关的脂肪肝病(MASLD),以前称为非酒精性脂肪肝病(NAFLD)1,是一种日益普遍的疾病,其特征是在无遗传性疾病的情况下出现肝脏脂肪变性。通过组织学检查可以将MASLD区分为良性状态或更值得关注的代谢功能障碍相关的脂肪性肝炎(MASH)1,2。如果不加以治疗,MASH可能发展为肝硬化、肝衰竭和肝癌(HCC)3。MASH是一种复杂的代谢综合征,由于对其发病机制的理解不足,治疗面临挑战。迄今为止,已有50多种在研新药针对各种代谢、炎症和纤维化相关的MASH靶点开发4。2024年3月,美国食品药品监督管理局(FDA)批准了口服甲状腺激素受体-β(THRβ)激动剂雷米特罗(Resmetirom)作为首个MASH治疗药物。在一项III期临床试验中,雷米特罗改善了约25-30%患者的肝脏病理状况5。未来治疗标准的改进可能需要联合疗法,这使得有必要开发其他类型的MASH治疗药物。
亲环素(Cyclophilins)是一类在人类生物学中起关键作用的蛋白质家族,通过协助蛋白质折叠和转运,参与调控多种细胞过程6,7。由于亲环素A亚型(Cyp A)在介导炎症中的作用以及亲环素D亚型(Cyp D)在坏死性细胞死亡中的作用,亲环素抑制剂被认为是具有吸引力的MASH治疗药物8-9。在MASH中,药理学抑制亲环素B亚型(Cyp B)可能尤为重要,因为已有的研究表明该亚型在胶原蛋白生成中发挥了作用10。之前的研究表明,与正常组织相比,纤维化组织中Cyp B的表达显著上调。纤维化是唯一与MASH患者肝衰竭显著相关的组织学标志物11。人们发现源自天然产物的非免疫抑制性亲环素抑制剂环孢素(Cyclosporine,CsA)或桑夫林(Sanglifehrin,SfA)12(12) 在肝纤维化的CCL4模型和MASH的小鼠模型中具有抗纤维化作用13-14。这些发现推动了非免疫抑制性亲环素抑制剂Rencofilstat的II期MASH临床试验15。
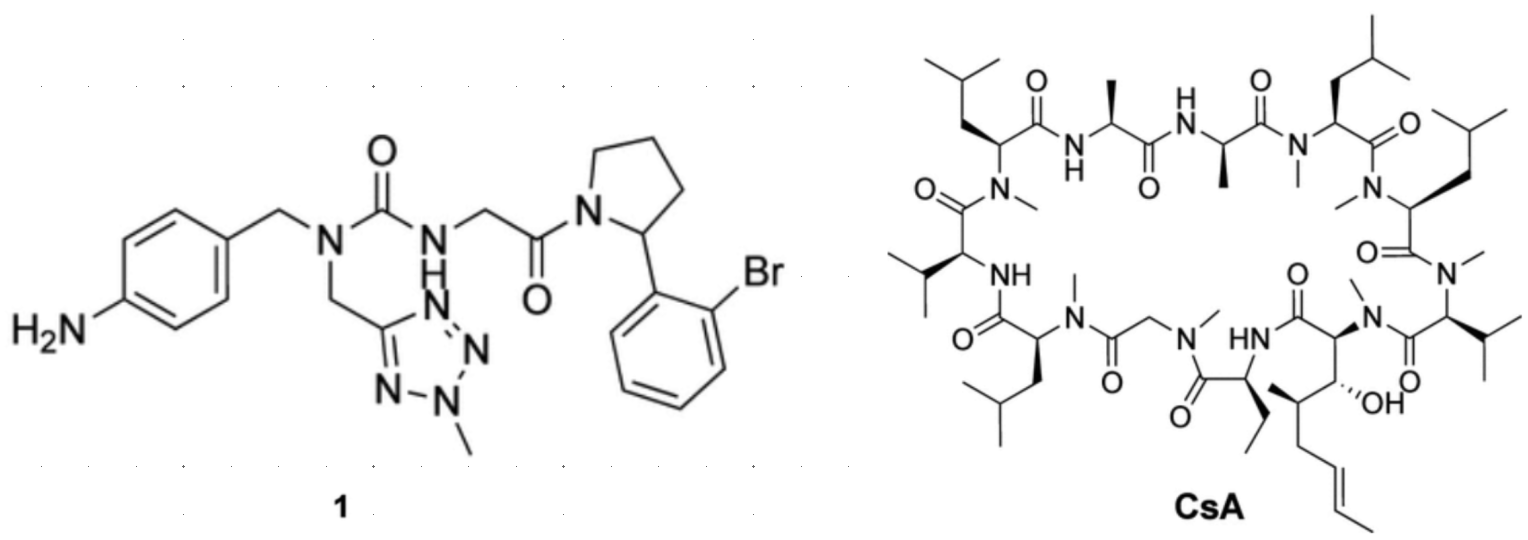
图1. 三臂亲环素抑制剂1和环孢素A(cyclosporine A ,CsA)的化学结构
目前已进入临床研究的亲环素制剂均为泛选择性(pan-selective)抑制剂,由于亲环素A的相对丰度较高,这可能会限制对亲环素B或D亚型的作用16-17。因此,开发具有更高亚型选择性且易于优化的新一类亲环素抑制剂是十分必要的,因为源自天然产物CsA或Sfa的亲环素抑制剂难以通过修饰来优化其药物特性。已有多个研究团队报道了各类小分子抑制剂18。由于大多数已知抑制剂所占据的两个主要口袋高度保守,实现亚型选择性通常颇具挑战性。Peterson等人16最近报道了一种基于大环骨架的Cyp D选择性抑制剂,但不得不依赖前药策略来实现细胞渗透性。我们19之前报道了化合物1(图1),一种小分子三臂亲环素抑制剂,它能够同时作用于亲环素表面的三个子口袋。在抗增殖细胞实验中,化合物1表现出与CsA相当的活性,并且细胞毒性较低,表明其可能与大环类亲环素抑制剂有所区别。然而,化合物1对Cyp B相较于Cyp A的选择性仅略有提高,并且含有一个被认为存在开发隐患的伯芳香胺基团,因为伯芳香胺可能经代谢活化为反应性氮鎓离子,与DNA形成共价加合物20。因此,我们着手设计改进型三臂亲环蛋白抑制剂,以解决致突变问题,优化亚型选择性和成药性。这些努力最终促成了化合物11的发下,这是一种强效的Cyp B抑制剂,具有令人鼓舞的药代动力学特性,适合进一步开发。
结果与讨论
分子模拟
首先,基于化合物1与Cyp A复合物的X射线衍射晶体结构(图2)构建了一个计算亲和力模型,并针对一系列先前报道的环孢素A苯基-吡咯烷脲配体进行了基准测试19,21。
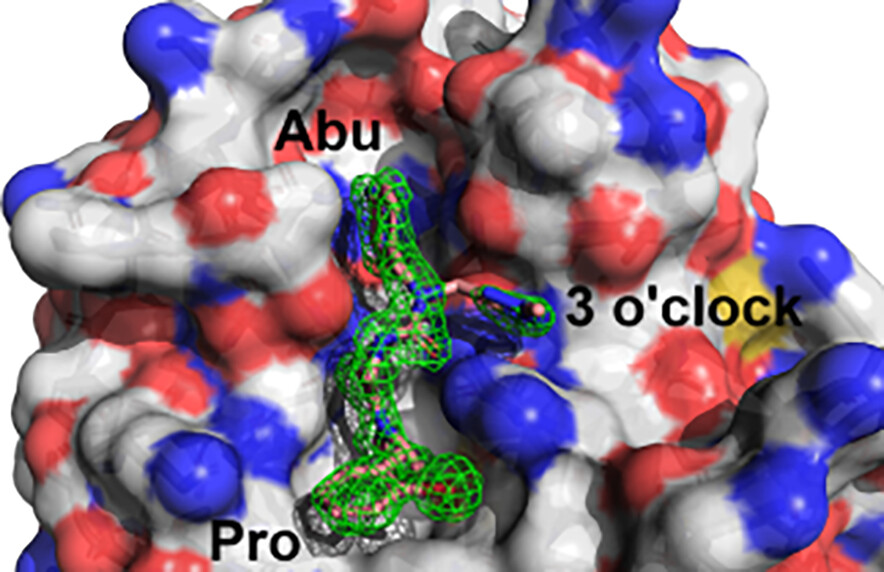
图2. 用于亲和力计算的Cyp A/1的X-衍射结构(PDB ID 6GJN)。化合物1占据了活性位点的Abu口袋、Pro口袋以及3点钟方向的辅助口袋。Fo – Fc电子密度差图以1.5σ等值图显示为绿色网格。
在基准测试中,采用Flare V5软件的Flare FEP (Flare V5)软件计算结合自由能22,所得的FEP亲和力模型表现出令人鼓舞的排序能力(图 S1):r² = 0.56,MUE = 0.4 kcal·mol⁻¹,Kendall τ = 0.67,并将该模型用于指导设计结构新颖且性能更优的抑制剂。
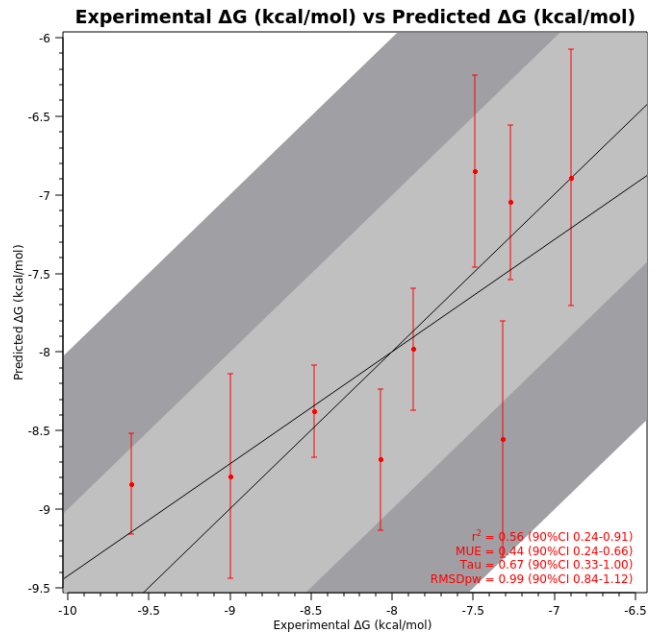
图 S1. 针对文献报道的9个CypA抑制剂数据集的亲和力模型。
因此,从化合物1出发,分别对Abu、Pro和3点钟方向的子口袋(图2)进行了FEP扫描分析。
在Pro子口袋方向优化的FEP模拟
对Pro口袋的修饰聚焦于用其他基团替换邻溴苯基吡咯烷取代基,以提高溶解性和亲和力。用氯或线性、支链、环烷基取代均导致亲和力下降,仅硫甲基化被预测可增强活性(Table S1)。
Table S1. 在Pro子口袋方向优化的FEP模型
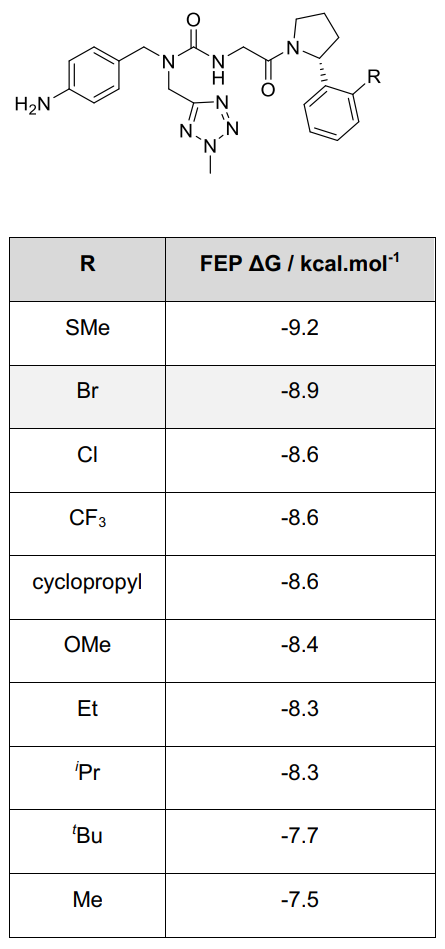
其中先导化合物1以灰色突出显示,结合亲和力估算值的统计不确定性约为±0.5 kcal·mol⁻¹。
在Abu子口袋方向优化的FEP模拟
在Abu口袋的FEP扫描则聚焦于克服与苯胺基团相关的遗传毒性问题。将氨基替换为羟基或酰胺基,以及进行甲基化或环化,均被预测会显著削弱亲和力。对Abu口袋的详细检查表明,氨基与Cyp A残基及一个埋藏的水分子形成了两个氢键。由于未能找到该基团的替换物,策略转向通过邻位卤代或用富含氮的杂环取代苯基来破坏1的氮鎓离子衍生物的稳定性23,24。这一方法得到了我们先前发现的支持,即氯化吡啶片段能够结合到Cyp A的Abu口袋中25。单卤化被预测为可适度提高亲和力,但二卤化降低了亲和力。吡啶或嘧啶的替换也被预测可增强亲和力。然而,对氨基吡啶变体的pKa预测表明,在生理条件下会有显著解离,这对结合到Abu口袋可能是不利的(Tbale S2)。
Table S2. 在Abu子口袋方向优化的FEP模型
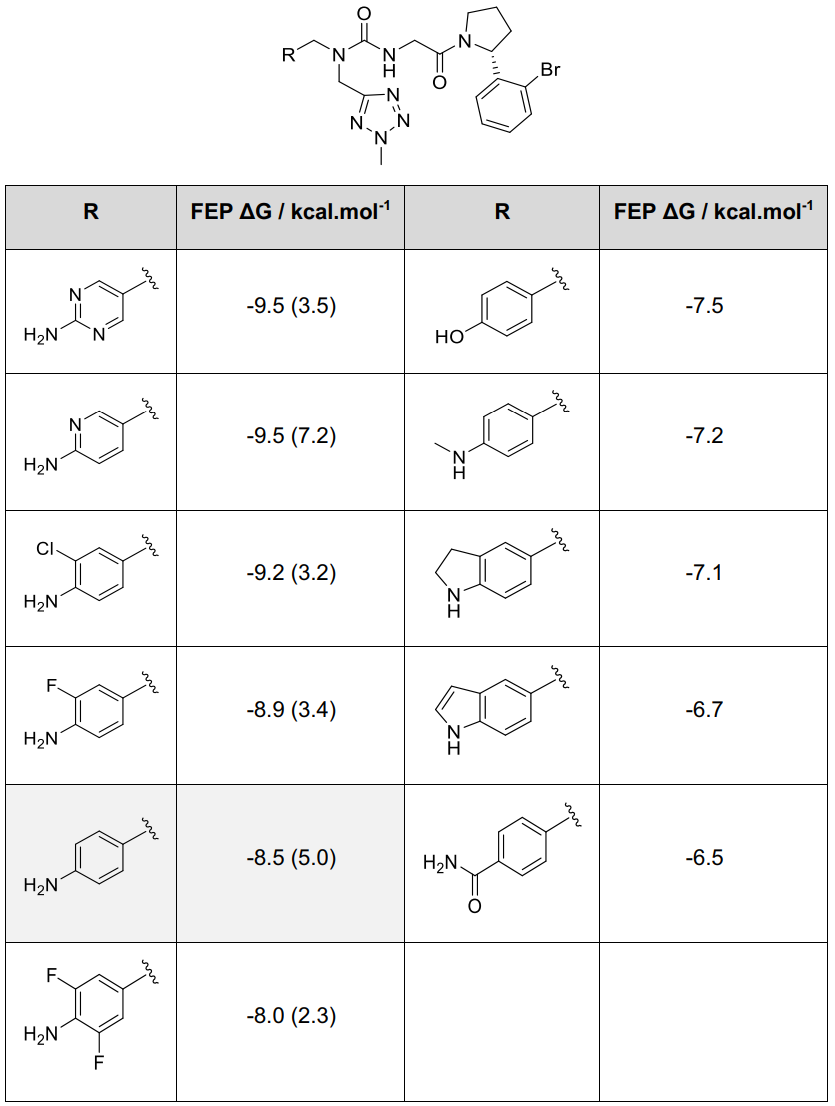
其中,括号中的值是用Chemicalize模型预测的R基团最强碱性pKa估算值。先导化合物1以灰色突出显示,化合物1的FEP亲和力估算值与表S1中的数据略有不同,因为它是基于一组不同的相对结合自由能估算值计算得出的。结合亲和力估算值的统计不确定性约为±0.5 kcal·mol⁻¹。
在3点钟子口袋方向优化的FEP模拟
在3点钟方向口袋的FEP扫描则聚焦于探索其他五元环的替换方案。用噻二唑、噻唑和噁二唑环替换四唑基团具有良好的耐受性。相比之下,呋喃、噁唑、异噁唑、三唑和噻吩变体则被预测为会导致中等程度的亲和力损失。这些修饰伴随着预测LogP值的显著变化,为平衡细胞渗透性和溶解性提供了空间(Table S3)。
Table S3. 在3点钟子口袋方向优化的FEP模型
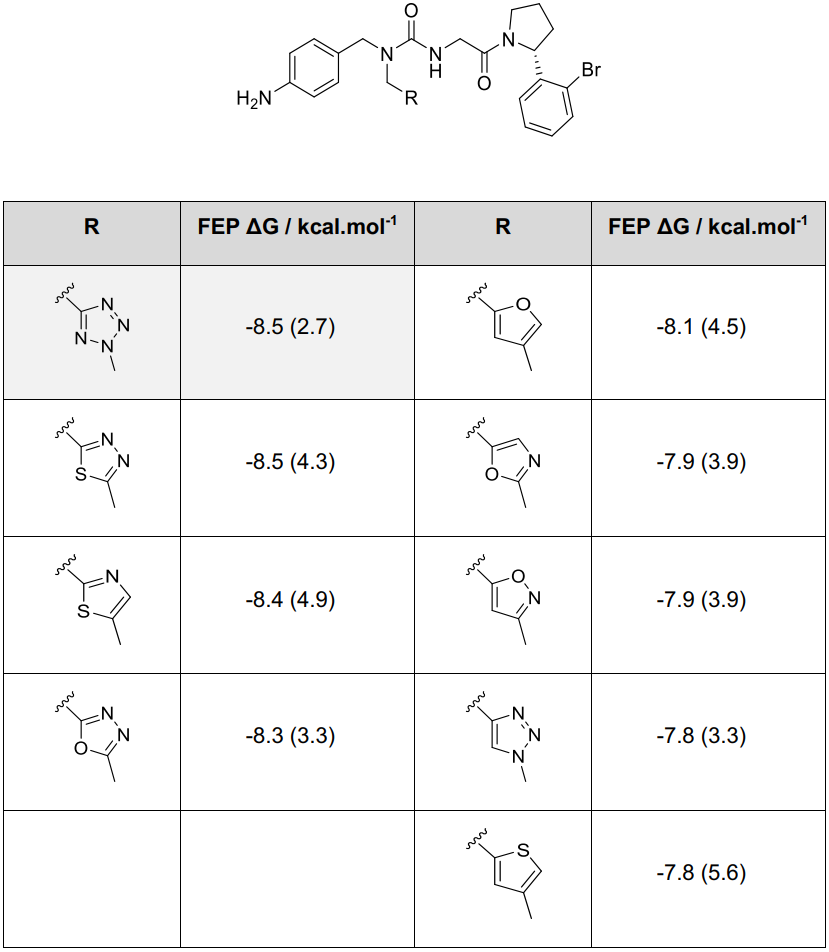
括号中的值是用Wildman-Crippen模型计算得到的cLogP值。先导化合物1以灰色突出显示,化合物1的FEP亲和力估算值与表S1中的数据略有不同,因为它是基于一组不同的相对结合自由能估算值计算得出的。结合亲和力估算值的统计不确定性约为±0.5 kcal·mol⁻¹。
组合设计
通过单独的FEP扫描识别出最有前景的设计特征,并将其组合,得到一系列化合物,这些化合物被预测为具有改进的结合特性,同时其cLogP值介于2到5之间(Table S4)。
Table S4. 具有改进的FEP结合亲合力预测值和cLogP值(约2-5)的化合物,用于合成优先级排序
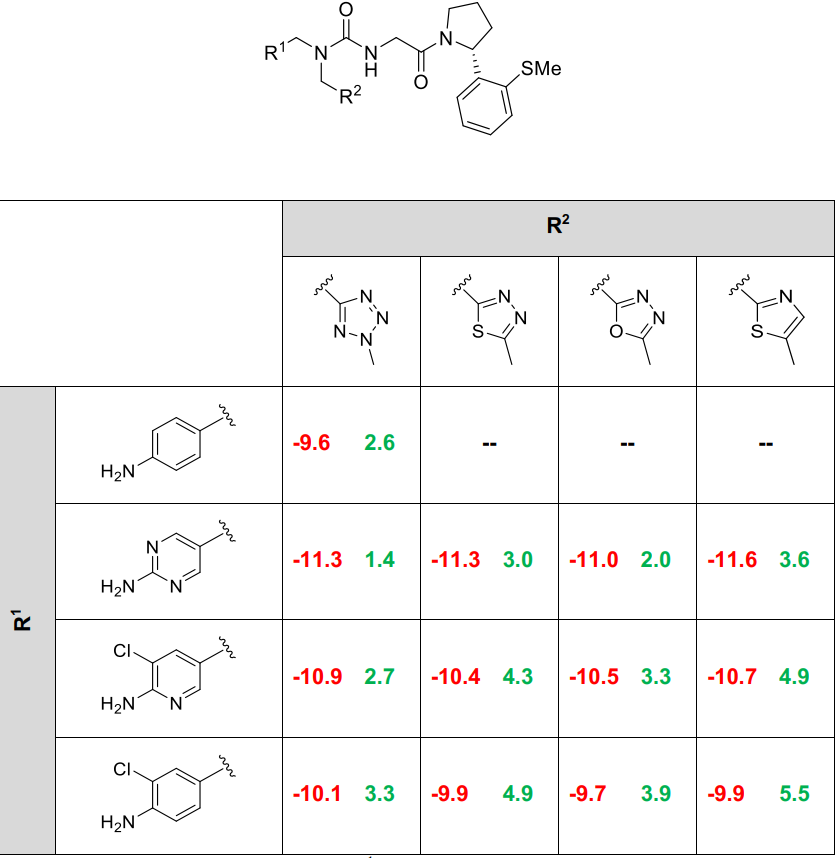
FEP计算值以红色显示(kcal·mol⁻¹),cLogP值以绿色显示。左上角化合物的FEP亲和力估算值与表S1中的数据略有不同,因为它是基于一组不同的相对结合自由能估算值计算得出的。结合亲和力估算值的统计不确定性约为±0.5 kcal·mol⁻¹。
计算靶标结合选择性分析
在基准模式下使用Flare FEP(Flare V8)计算化合物1、2、5-14的相对结合自由能。以1/Cyp A的X-衍射晶体结构(PDB 6GJN)为起点,手动建模所有类似物。通过将Cyp B和Cyp D的X-衍射结构1CYN(Cyp B)和4J5B(Cyp D)与Cyp A模型叠合,并在删除与配体冲突的晶体水分子后进行一轮能量最小化,构建了Cyp B和Cyp D的模型。
在所有FEP计算中,配体使用OpenFF 2.1.0进行参数化,并通过DFT/GFN2-xTB方法自定义扭转角参数。在平衡过程中,使用GCNCMC优化配体周围的水分子位置。中间体被自动添加到计划的RBFE网络中,以最大化成对转换中所有分子的相似性。所有边均进行双向处理。对于导致循环闭合不佳的噪声边,重复计算并添加额外的边至RBFE网络,以降低结合亲和力估算的整体不确定性。
用于选择性分析的最终计算结合亲和力如表S5所示,Flare FEP计算输入的3D模型可从支持信息获取。
Table S5. 化合物1、2、5-14计算的结合自由能
| Comp. | CypA ΔG | Uncertainty | CypB ΔG | Uncertainty | CypD ΔG | Uncertainty |
|---|---|---|---|---|---|---|
| 1 | -7.1 | 0.5 | -8.4 | 0.7 | -7.4 | 0.3 |
| 2 | -8.5 | 0.3 | -9.4 | 0.4 | -8.5 | 0.1 |
| 5 | -9.2 | 0.3 | -10.3 | 0.3 | -9.7 | 0.1 |
| 6 | -7.7 | 0.4 | -6.7 | 0.6 | -5.8 | 0.2 |
| 7 | -6.8 | 0.9 | -6.8 | 1.0 | -6.1 | 0.2 |
| 8 | -8.3 | 0.5 | -8.3 | 0.6 | -8.2 | 0.1 |
| 9 | -8.3 | 0.7 | -8.3 | 0.5 | -8.8 | 0.4 |
| 10 | -8.5 | 0.5 | -9.2 | 0.6 | -8.6 | 0.1 |
| 11 | -9.8 | 0.3 | -11.1 | 0.4 | -10.0 | 0.4 |
| 12 | -9.5 | 0.5 | -11.3 | 0.6 | -9.6 | 0.2 |
| 13 | -7.9 | 0.5 | -11.5 | 0.6 | -10.0 | 0.2 |
| 14 | -9.1 | 0.4 | -10.2 | 0.5 | -9.5 | 0.1 |
单位:kcal/mol
合成、活性测试与SAR分析
通过表面等离子体共振(SPR)技术评估了这些化合物与亲环素Cyp A、B和D亚型的的结合特性。表1列出了三个方向的取代基结构与或活性,代表性SPR数据见图3A。此外,还对化合物11和CsA进行了等温滴定量热法(ITC)实验,为SPR测得的Kd值提供了正交验证。
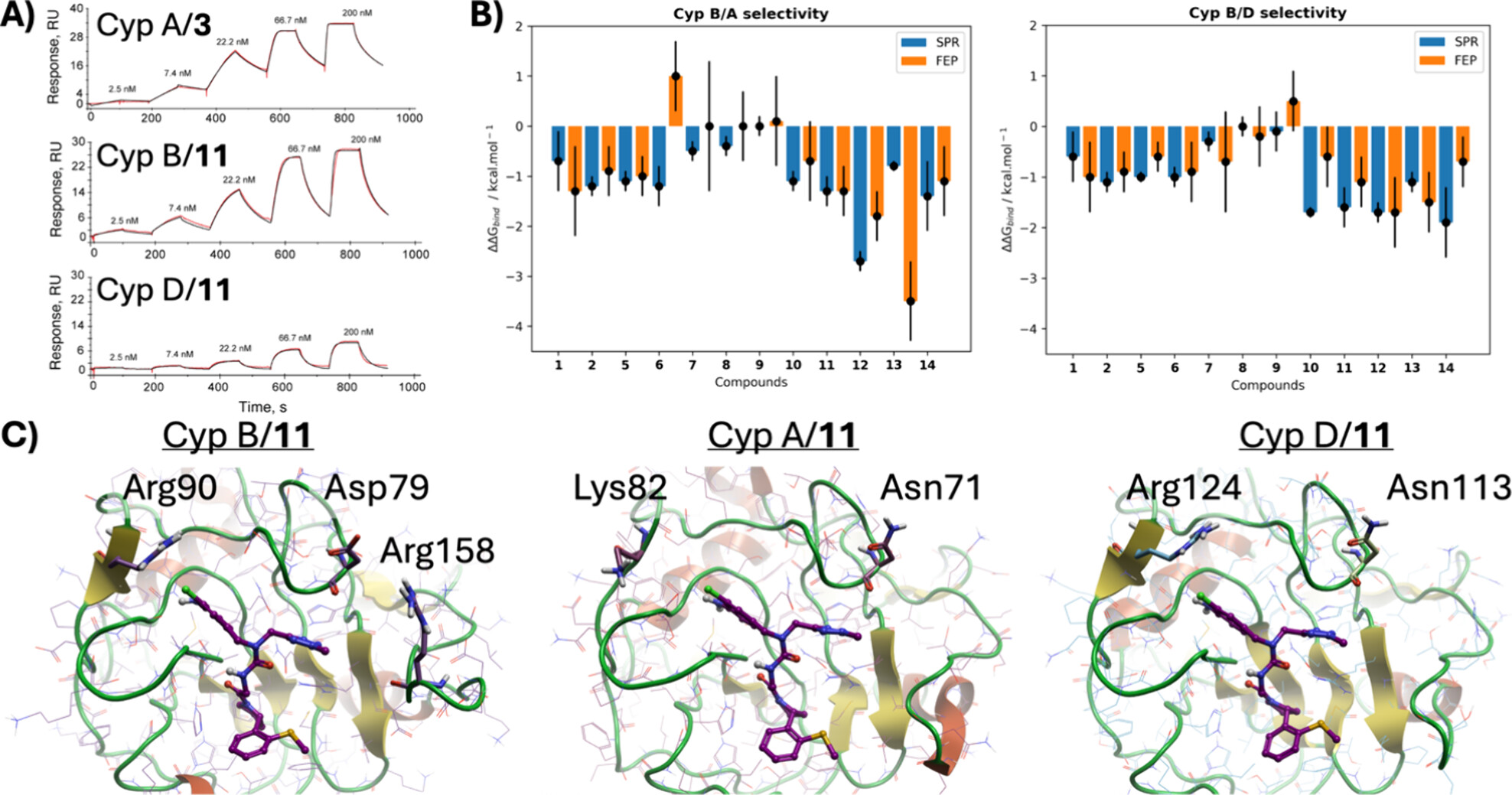
图3. A)表面等离子体共振(SPR)动力学滴定实验的代表性传感图。B)通过SPR测量或FEP计算得到的Cyp B/Cyp A和Cyp B/Cyp D之间的结合选择性谱的比较。负值表示优先与Cyp B结合。C)分子动力学(MD)模拟的代表性快照,展示了化合物11与Cyp B、Cyp A、Cyp D结合的模型。Abu口袋和3点钟方向口袋中在三种亚型之间不保守的残基以球棍模型表示。模型分别基于PDB ID 6GJN(Cyp A)、1CYN(Cyp B)、4J5B(Cyp D)构建。
将Pro口袋的溴苯基团替换为硫甲基苯基(1→2)使三种Cyp亚型的结合亲和力提高了10至20倍,与FEP预测一致。将Abu口袋的苯胺基团进一步替换为Gräedler等人36报道的并环四氢吡喃(2 → 3),又使亲和力提高了10倍。化合物3对Cyp B表现出令人印象深刻的低Kd值(2 nM),尽管对Cyp A的选择性仅略高(3倍)。将(R)-苯基吡咯烷替换为其(S)-对映体后,活性下降了10至40倍(3 → 4)。FEP模型表明,通过二氟化策略使苯胺失活(2 → 5)会导致约3倍的亲和力损失,但SPR测试显示,所有三种亚型的亲和力损失更为显著(约10至15倍)。在3点钟方向口袋中,将四氮唑替换为三氮唑导致了进一步的、符合FEP估算的3至4倍亲和力损失(5 → 6)。将四唑替换为较小的线性炔基(5 → 7)则无法容忍,活性显著下降。这些结果证实了占据3点钟方向口袋对于在该系列化合物中实现具有竞争力活性的重要性。通过将甲基取代的四唑替换为环戊基或环己基取代的四唑(1 → 8 和 1 → 9),进一步评估了3点钟方向口袋深处的扩展。这些修饰并未显著改善或削弱亲和力,但以显著增加的cLogP值为代价。
表1. 化合物1-14用SPR方法测得的解离常数Kd
| Compound | Cyp A | Cyp B | Cyp D |
|---|---|---|---|
| CsA | 22±4 (32±20) |
20±3 | 14±5 |
| 1 | 600±400 | 200±100 | 520±100 |
| 2 | 90±10 | 12±4 | 80±10 |
| 3 | 6±2 | 2±1 | 8±3 |
| 4 | 310±40 | 420±80 | 400±30 |
| 5 | 1160±200 | 200±40 | 1010±120 |
| 6 | 4600±2400 | 630±100 | 2970±800 |
| 7 | 8750±2900 | 3740±780 | 5850±2000 |
| 8 | 150±20 | 80±20 | 80±10 |
| 9 | 320±70 | 310±170 | 360±200 |
| 10 | 200±40 | 35±6 | 580±100 |
| 11 | 110±50 (80±6) |
12±3 | 180±100 |
| 12 | 7300±1000 | 88±20 | 1500±500 |
| 13 | 670±100 | 184±7 | 1200±300 |
| 14 | 960±130 | 92±75 | 2400±430 |
单位:nM;括号里的值是ITC分析结果。
这些努力促使我们在3点钟方向口袋中保留一个修饰最少的五元环,并探索其他策略以摆脱苯胺基团带来的潜在问题。在苯胺氨基邻位引入单氯化修饰使得活性提高了3至5倍,这与FEP模型预测一致(1 → 10)。这一修饰与溴基到硫甲基苯基吡咯烷的替换相结合,得到了化合物11,其对Cyp B表现出高亲和力(Kd约为10 nM),并对Cyp A和Cyp D显示出10至20多倍的选择性。
随后,我们尝试用嘧啶和氯吡啶变体替代苯胺,同时结合四唑替换以将cLogP值控制在3到4的范围内。基于这些考虑,我们制备了含噁二唑、噻唑或噻二唑杂环占据3点钟方向口袋的变体。与预测的FEP趋势不符的是,含有嘧啶和氯吡啶Abu口袋变体的噻唑和噻二唑系列(12、13、14)相较于化合物11表现出较弱的亲和力。令人惊讶的是,化合物12对Cyp B相对于Cyp A表现出高达80倍的选择性。所有乙内酰脲衍生物(15、16、17、18、19)相较于基于线性脲的化合物,对所有亚型的亲和力均显著下降(表2)。这可能是由于吡咯烷基团附近的脲氮参与的氢键相互作用被破坏所致。
表2. 化合物15-19用SPR方法测得的解离常数Kd
| Compound | Cyp A | Cyp B | Cyp D |
|---|---|---|---|
| 15 | 6700±2600 | 3800±1300 | 6300±1600 |
| 16 | 7100±1250 | 4500±500 | 9100±900 |
| 17 | \(>\)10,000 | \(>\)10,000 | \(>\)10,000 |
| 18 | \(>\)10,000 | \(>\)10,000 | \(>\)10,000 |
| 19 | \(>\)10,000 | \(>\)10,000 | \(>\)10,000 |
单位:nM
为进一步研究观察到的结合选择性趋势,使用Flare FEP(Flare V8)进行了另一轮FEP模拟。因此,模拟了化合物1、2、5-14与Cyp A、Cyp B和Cyp D的复合物。化合物3和4因涉及并环四氢吡喃环的炼金术转换在技术上具有挑战性而被排除。化合物15-19由于其亲和力较差(表2)也被排除。图3B展示了通过SPR测量和FEP计算得到的Cyp B/A和Cyp B/D结合选择性比率的比较。尽管对于所有化合物,SPR测量与FEP计算之间没有定量的一致性,但两种方法在总体上均一致表明,该配体系列对Cyp B的优先结合高于其他两个亚型。结合选择性的差异可以通过Abu口袋和3点钟方向口袋中残基的差异来解释(图3C)。Cyp B中的Arg90在Cyp A中被Lys82取代,导致Cyp A的口袋更不封闭。Cyp B中的Asp79在Cyp A中被Asn71取代,在Cyp D中则被Asn113取代。此外,Cyp B中的Arg158是在与Cyp A和Cyp D序列比对中的插入位点。这些差异导致了位于3点钟方向口袋中的与杂环静电相互作用的变化。
细胞实验
随后,我们通过在透化处理的人肝癌细胞(HepG2)中使用钙保留能力(Calcium Retention Capacity)实验,寻找一些合成化合物在细胞环境中与亲环素相互作用的证据。HepG2是一种常用于体外MAFLD研究的细胞系37。值得注意的是,在钙保留实验中,化合物3表现出比环孢素(CsA)高出2倍的活性(EC50值分别为244 nM和463 nM,图4A)。四种测试化合物的SPR测定的Cyp D解离常数与测得的钙保留实验EC50值之间呈现出强烈的相关性(图4B)。这些结果表明,这些化合物可能通过抑制Cyp D介导的线粒体通透性转换孔(mitochondrial permeability transition pore)在钙过载条件下的开放而发挥作用38。
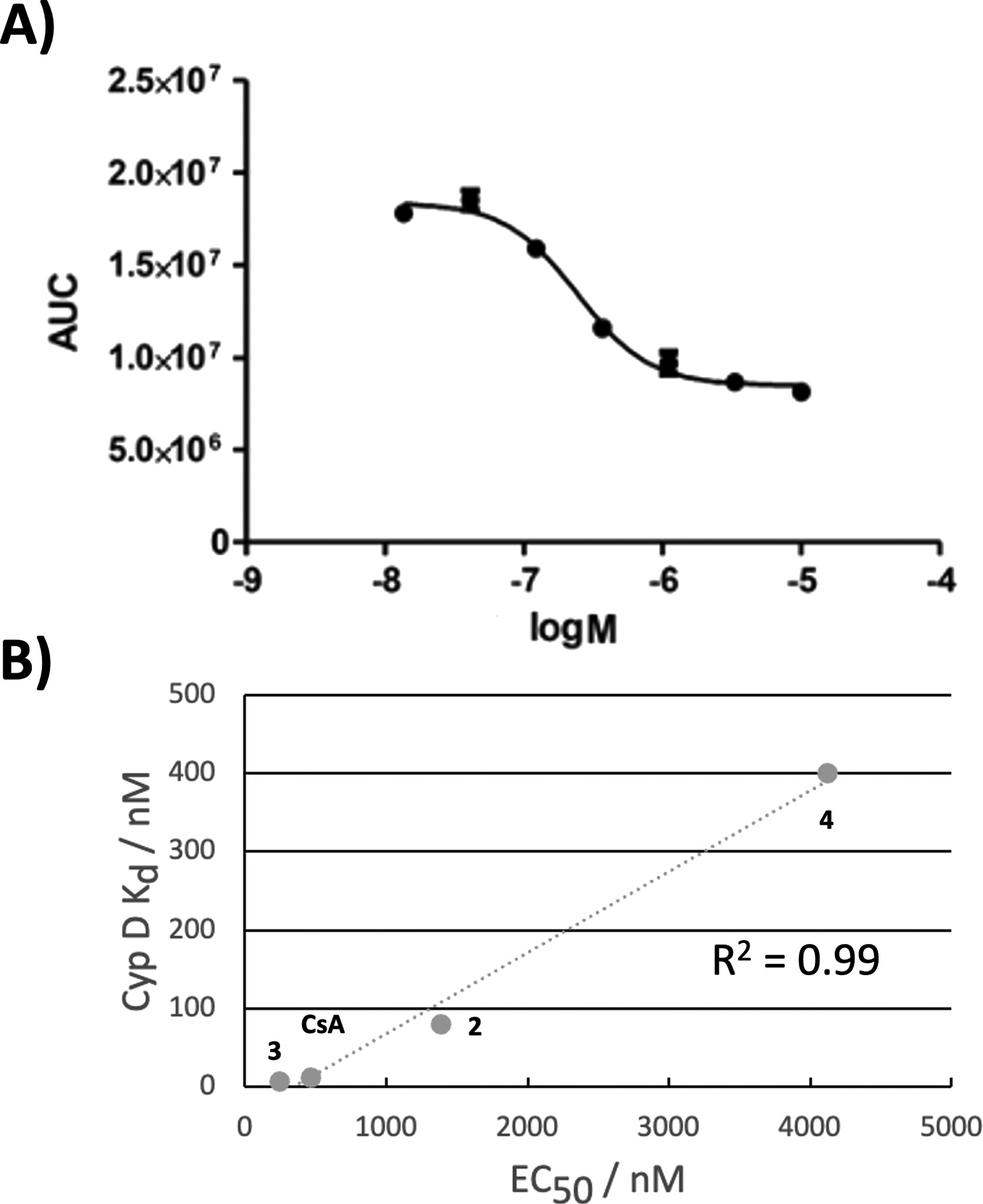
图4. HepG2细胞中的钙保留能力实验。A) 化合物3的浓度响应曲线。B) 四种化合物的EC50值与SPR测定的Cyp D解离常数之间的相关性。
我们还寻找了精选化合物在治疗脂肪肝疾病方面的有效性证据。最近的一项研究表明39,在化学和饮食诱导的MASH(代谢相关脂肪性肝病)小鼠模型中,敲除Cyp B显著保护了肝脏免受脂质积累和纤维化的影响,而敲除Cyp A则没有此效果。使用诱导多能干细胞(iPSC)衍生的肝细胞——这些细胞在加载游离脂肪酸时表现出与MASH表型一致的基因型和表型特征——我们评估了用Cyp B选择性化合物11和12处理48小时后的脂肪变性情况,并将其与非选择性CsA的效果进行了比较。化合物11对Cyp B的亲和力比化合物12高出约7倍,表现出良好的活性,显著降低了肝细胞的整体脂质水平(图5A、B)以及脂滴大小(图5C),而CsA则没有这种效果。有趣的是,在这项实验中,这种降低程度与FDA批准的MASH治疗药物Resmetirom(Res)所诱导的效果相当(图5A、5D);两者的EC50值相似(约为0.3 μM)。然而,Resmetirom似乎能够比化合物11更大幅度地降低脂质水平。Resmetirom的EC50曲线底部更低(图5A),每细胞约11 μm²,而化合物11为36 μm²,使用10 μM浓度处理后,整体细胞脂质水平(图5B)以及平均脂滴大小(图5C)也低于化合物11。据我们所知,三臂选择性Cyp B抑制剂和Resmetirom的作用机制是独立的,因此它们有可能联合使用以提高疗效。
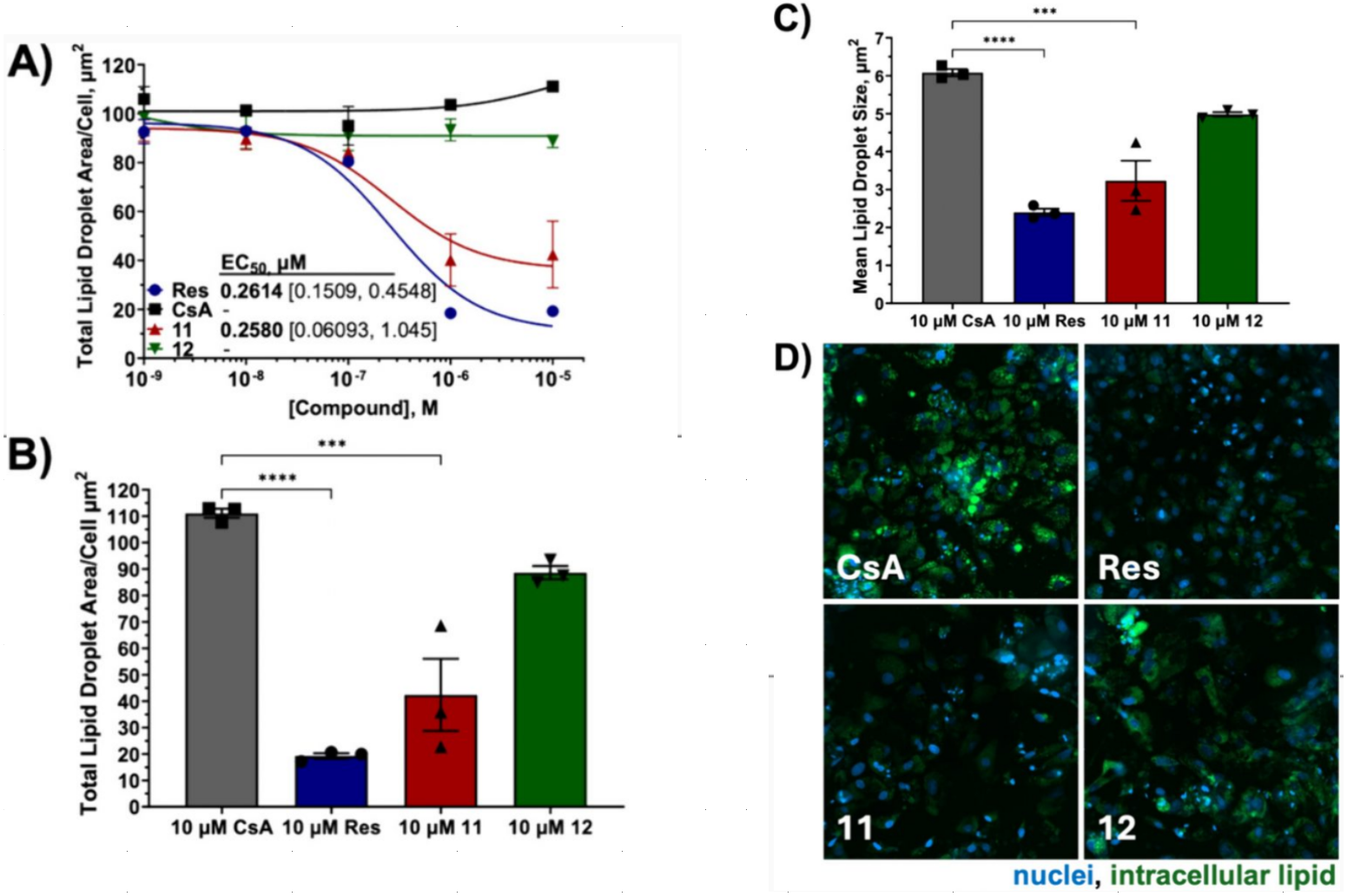
图 5. 化合物对 48 小时后肝细胞脂肪变性的影响。A)CsA、Resmetirom (Res)、11 和 12 处理后的脂肪变性浓度反应曲线;EC50 及其 95% CI [下限,上限]。B)每个细胞的肝细胞脂肪变性总面积。C)平均脂滴大小。D)代表性图像 1 mm × 1 mm。A-C 图中的数值为 3 次独立实验的平均值 ± SEM,每次实验有 6 个重复;**** p \(<\) 0.0001,*** p \(<\) 0.001。
DMPK与毒性
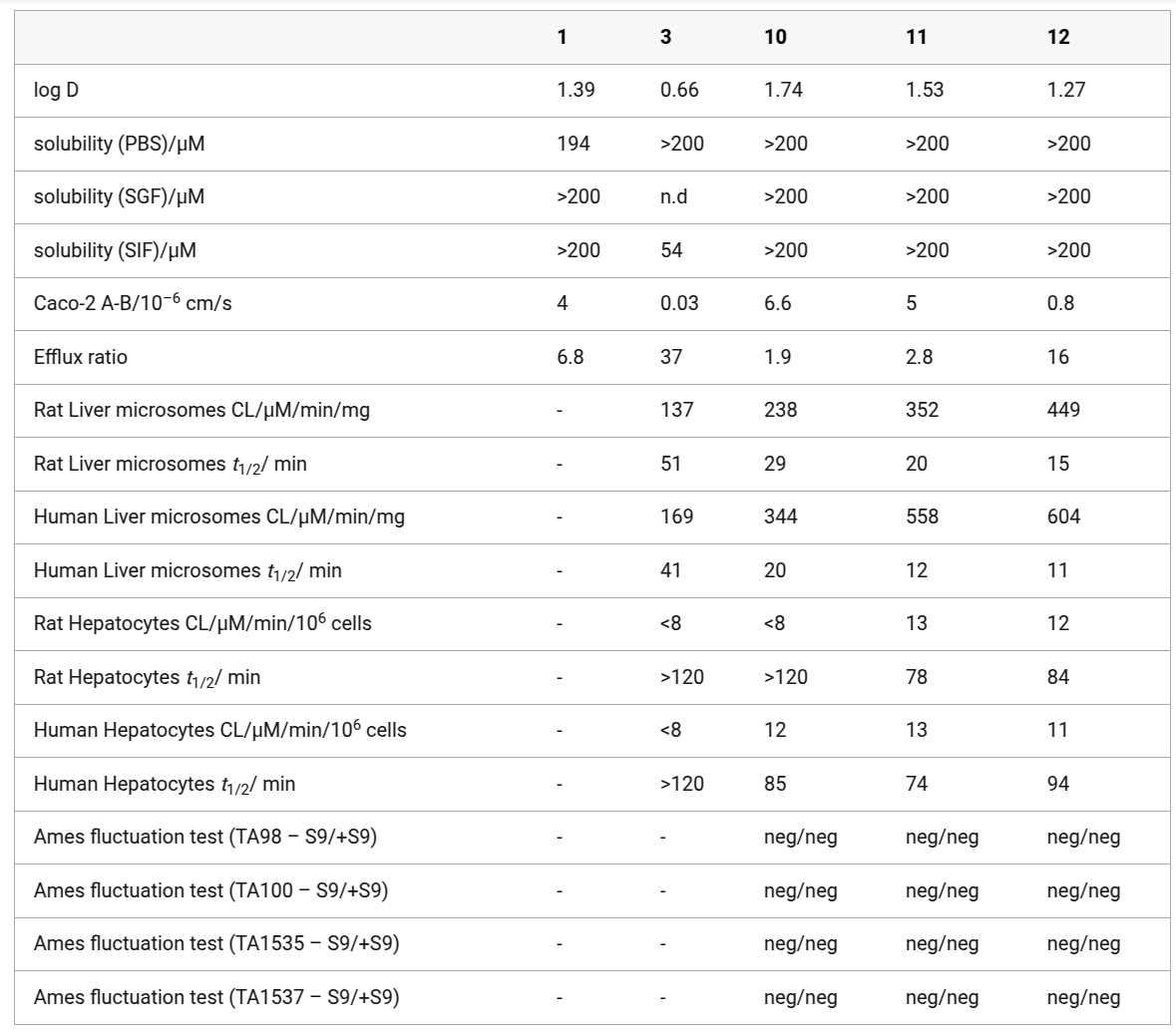
对化合物1、3、10、11和12的体外代谢进行了评估,以确定该系列化合物进一步开发的潜力(表3)。水/辛醇分配系数(Log D)被认为在可接受范围内。所有化合物在pH 7.4时均具有很高的溶解性,但化合物3在模拟肠液(SIF)中的溶解性显著降低。化合物3在模拟胃液(SGF)中无法被检测到,这表明在酸性条件下,环化四氢吡喃部分可能存在化学不稳定性。化合物1显示出令人鼓舞的Caco-2表观渗透性,但外排比很高。化合物10和11在保持与化合物1相似的表观渗透性的同时,降低了外排比。相比之下,化合物3和12表现出显著降低的Caco-2表观渗透性和增加的外排。化合物12的低表观渗透性和高外排可能是其在MASH肝细胞实验中缺乏效力的原因。相反,在钙保留能力实验中,化合物3可能表现出较强的活性,因为HepG2细胞在处理前用洋地黄皂苷进行了通透化处理。所有化合物在人和大鼠肝微粒体中都显示出高清除率。化合物3在人和大鼠肝细胞中表现出低清除率,这与其低被动渗透性一致。化合物10、11和12在人和大鼠肝细胞中表现出中等清除率。这些结果表明,虽然化合物3的环化四氢吡喃片段赋予了其优异的亲环素结合亲和力(表1),但也使母体化合物无法进入细胞。化合物10-12在肝微粒体中的清除率比在肝细胞中更快,这可能是CYP代谢的一个迹象40。令人欣慰的是,通过卤代(10、11)或用嘧啶取代苯基(12)来减轻苯胺部分基因毒性的策略被认为是成功的,因为这三个化合物在所有测试菌株中,无论是否添加肝S9组分,均为Ames阴性。
表3. 一组化合物的DMPK和致突变性数据
进一步的特性分析(表4)表明,化合物11对细胞色素P450酶系表现出中等(CYP2C8、CYP2C19、CYP3A)或较弱(CYP1A、CYP2B6、CYP2C9、CYP2D6)的抑制作用,总体上显示出令人鼓舞的药物-药物相互作用特性。化合物11在大鼠和人血浆蛋白结合实验中均进行了测定,在研究的时间过程中未发现血浆稳定性问题。
表4. 化合物11的血浆蛋白结合率和细胞色素P450抑制测定结果a
| Rat PPB free % | 11 | Human PPB free % | 2 |
| Rat plasma recovery % | 99 | Human plasma recovery % | 88 |
| CYP1A phenacetin % | 25 | CYP2B6 bupropion % | 11 |
| CYP2C8 amodiaquine % | 71 | CYP2C9 diclofenac % | 45 |
| CYP2C19 omeprazole % | 72 | CYP2D6 dextrometor. % | 18 |
| CYP3A midazolam % | 73 | CYP3A testosterone % | 59 |
a.在10 μM的单点测试结果
结论
亲环素(Cyclophilin)抑制剂为治疗MASH(代谢功能障碍相关脂肪性肝炎)及相关肝病提供了一种极具前景的潜在治疗策略。开发选择性作用于Cyp B而非Cyp A的抑制剂尤其引人注目,因为Cyp B在肝纤维化中发挥了重要作用。迄今为止,只有源自天然产物的非亚型选择性亲环素抑制剂进入了临床研究12,18。早期曾有报道显示某些小分子Cyp A抑制剂具有强效活性41,但其他研究团队未能重复这些结果21。Daum等人报道了芳基茚酮类抑制剂,这类化合物在PPIase酶活性测定中对Cyp A的选择性高于Cyp B,其解离常数为低微摩尔级别42,但正如本文所指出的,在MASH背景下更希望实现对Cyp B或Cyp D的选择性抑制,而非Cyp A。Petersson等人通过双苯基二羧酸酯大环化合物实现了强效且选择性的Cyp D抑制,但需要采用前药策略才能将化合物递送至细胞内,并且未报告相关的药物代谢和药代动力学(DMPK)数据16。Ahmed-Belkacem等人报道了一系列苯基吡咯烷脲类化合物,这些化合物在体外对Cyp A、Cyp B和Cyp D表现出强效活性,但未报告亚型选择性抑制21。Shore等人和Gräedler等人后来分别优化了这一系列化合物,得到了强效的Cyp D抑制剂,但未报告结合选择性或DMPK与致突变性数据以证明其在体内研究中的开发潜力36,43。这些努力突显了开发强效且亚型选择性的小分子亲环蛋白抑制剂所面临的挑战。在此,我们利用自由能计算优化了一类三臂亲环素抑制剂,这类抑制剂作用于一个保守性较低的辅助“3点钟方向”口袋。这些研究指导了化合物的合成,使其在表面等离子体共振(SPR)实验中对Cyp B达到了个位数nM级别的效力,并实现了高达80倍的Cyp B/A选择性。该系列化合物的细胞活性在人类HepG2细胞以及表现出MASH样特征的iPSC衍生肝细胞中得到了验证。四个强效类似物被推进到体外DMPK实验和致突变性实验。这些实验验证了为减轻遗传毒性风险而引入的理性化合物修饰。化合物11最终成为一种强效的Cyp B抑制剂,对Cyp A的选择性提高了10倍,同时表现出良好的药物安全性、体外DMPK特性以及降低肝脂肪变性的功效;而肝脂肪变性正是MASH疾病的关键组成部分。因此,这一化合物系列有望用于研究选择性Cyp B抑制在MASH及相关肝病治疗中的潜在益处。
联系我们
Flare V10交付先进的科学方法、分析工具和直观、易用的增强功能,洞察您的配体-蛋白质复合物结构。
想要尝试Flare信息丰富、用户友好界面,发现它如何帮助您自信地推动潜在先导化合物优化?请现在就联系我们安排试用,快速访问Flare的广泛功能。我们的专业团队随时准备通过安装和设置为您提供支持,而我们全面的教程库——涵盖从常见工作流程到高级方法和功能的所有内容将帮助您开始使用。我们在这里帮助您更快地实现目标,让您设计出感兴趣的分子。
- 电邮:info@molcalx.com
- 电话:020-38261356


