
摘要:本文总结了Flare 自由能微扰(Flare FEP)技术最近几年的关键进展。相对结合自由能(RBFE)虽在先导优化中表现优异,但受限于结构变化范围小。绝对结合自由能(ABFE)突破此局限,支持对全新分子独立计算,适用于早期虚拟筛选与广阔化学空间探索,并可灵活适配不同靶点构象与质子化状态。技术层面,FEP效率显著提升:自动λ调度算法优化计算窗口;GCNCMC与3D-RISM方法精准模拟水合环境;膜蛋白体系通过截断建模平衡精度与速度。主动学习策略融合高精度FEP与快速QSAR,实现“小样本精调—大范围预测”的迭代优化,大幅降低整体成本。尽管ABFE计算耗时约为RBFE的10倍,且存在系统性偏差(源于忽略蛋白质动态响应),但随着平台成熟与对构象变化理解加深,其潜力巨大。未来有望通过预测驱动方式,实现从虚拟筛选到候选分子优化的高效闭环,推动药物发现迈向数字化、精准化新阶段。
作者:Stuart Firth-Clark/August 27, 2025
前言
时光飞逝,转眼间已过去五年!我简直难以置信,距离我撰写博客文章——《自由能微扰(FEP):药物发现工具箱中的又一利器》——已经过去这么久。当时我介绍了FEP技术及其在进行此类计算时所需的一些核心概念。如今回望,我觉得有必要再次探讨FEP,因为这五年间,该领域发生了许多令人振奋的变化。
FEP依然是一个极具前景的研究方向。随着这一技术日益可靠且预测能力不断增强,它正展现出更大的潜力,帮助以科学为基础的产业——如制药、生物技术和农业化学行业——实现更高的效率。其核心优势在于,能够推动这些行业摆脱传统昂贵且耗时的“实验室探索”模式,转而采用更高效、更精准的计算机模拟预测方法。
随着我们对它的理解不断深入,FEP技术持续发展,其模拟策略也日趋精确。接下来,让我们回顾一下过去五年中FEP领域取得的一些重要进展。
λ窗口的选取
在设置FEP微扰路径时,一个关键挑战是确定每条连接(link)应计算多少个λ窗口。计算的窗口过少可能导致结果不准确;而窗口过多则会浪费宝贵的GPU算力资源。
五年前,我通常根据转化过程的复杂程度(即参与变化的原子类型和数量)来“猜测”所需的λ窗口数量。然而,这种凭经验的猜测常常出错,导致必须重新计算整个连接,既耗时又令人沮丧。
如今,通过运行“短时探索性计算”,可以自动获得一个更合理的、基于数据的“智能预估”,从而确定所需λ窗口的数量。这种方法不仅能有效降低GPU成本,还能减轻科研人员必须“猜对”的心理压力,让研究过程更加从容高效,真正实现科学探索中的“心中有数”。
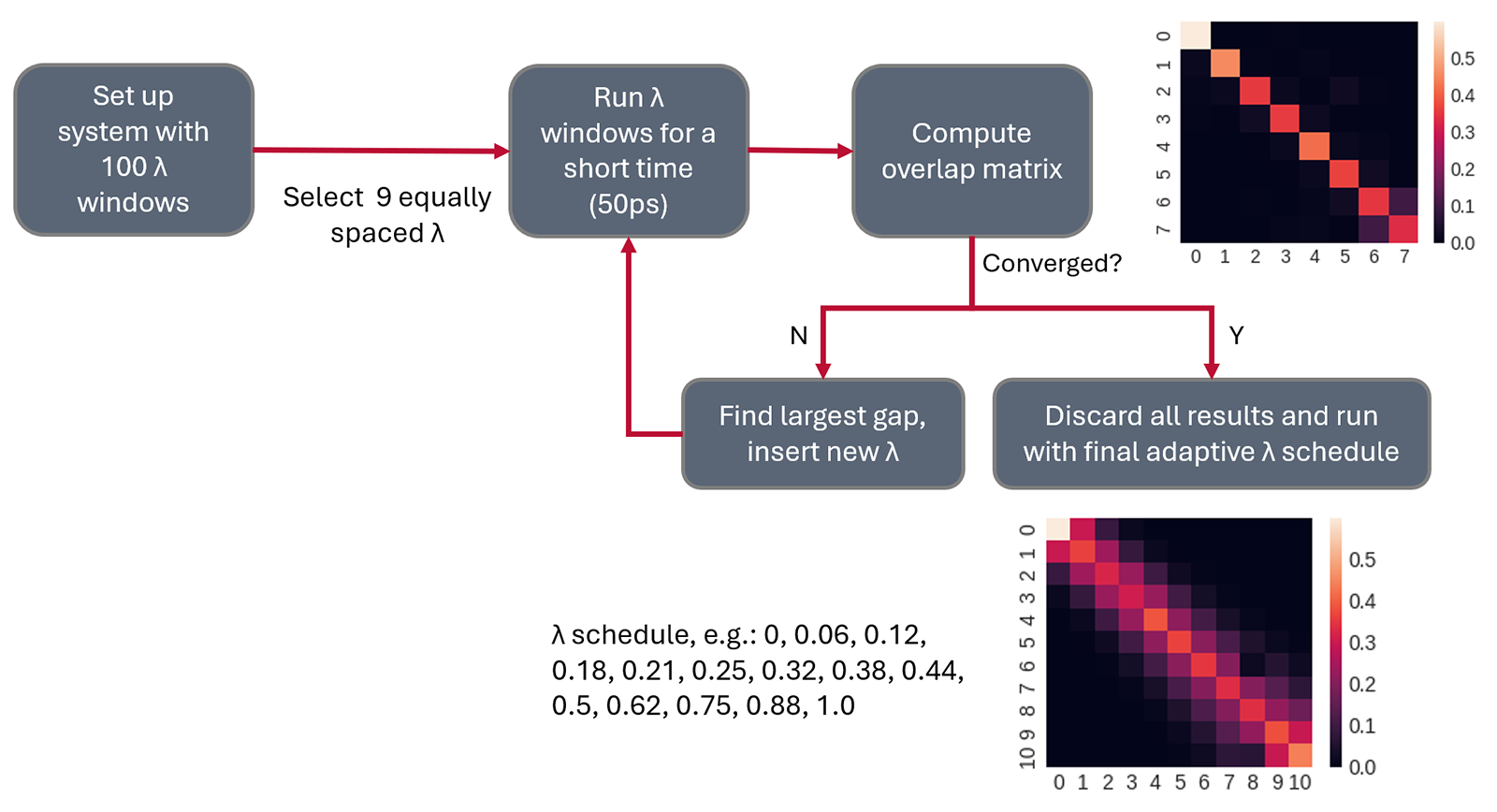
图1. 自动λ调度算法显著减少了在确定每个转化所需计算的λ窗口数量时所需的猜测工作量。
力场
任何FEP计算的核心在于对体系的描述与建模方式。准确地构建分子模型是获得可靠模拟结果的关键前提。如果分子体系仅包含“标准残基”以及在力场描述范围内的配体原子,那么其分子描述通常已足够精确,有望获得优异的计算结果。然而,现实中的自然体系和生物分子往往远比理想情况复杂得多!
对于配体等非标准体系,常常会因力场对体系的描述不足而产生显著误差,尤其是对二面角(torsion angles)的描述不准确时更为明显。这类问题在传统力场中尤为突出。一种可行的解决方案是通过量子力学(QM)计算,为特定的二面角生成更精确的参数。通过这种优化,可以显著提升分子体系的描述精度,从而进一步提高FEP模拟的准确性与预测能力。
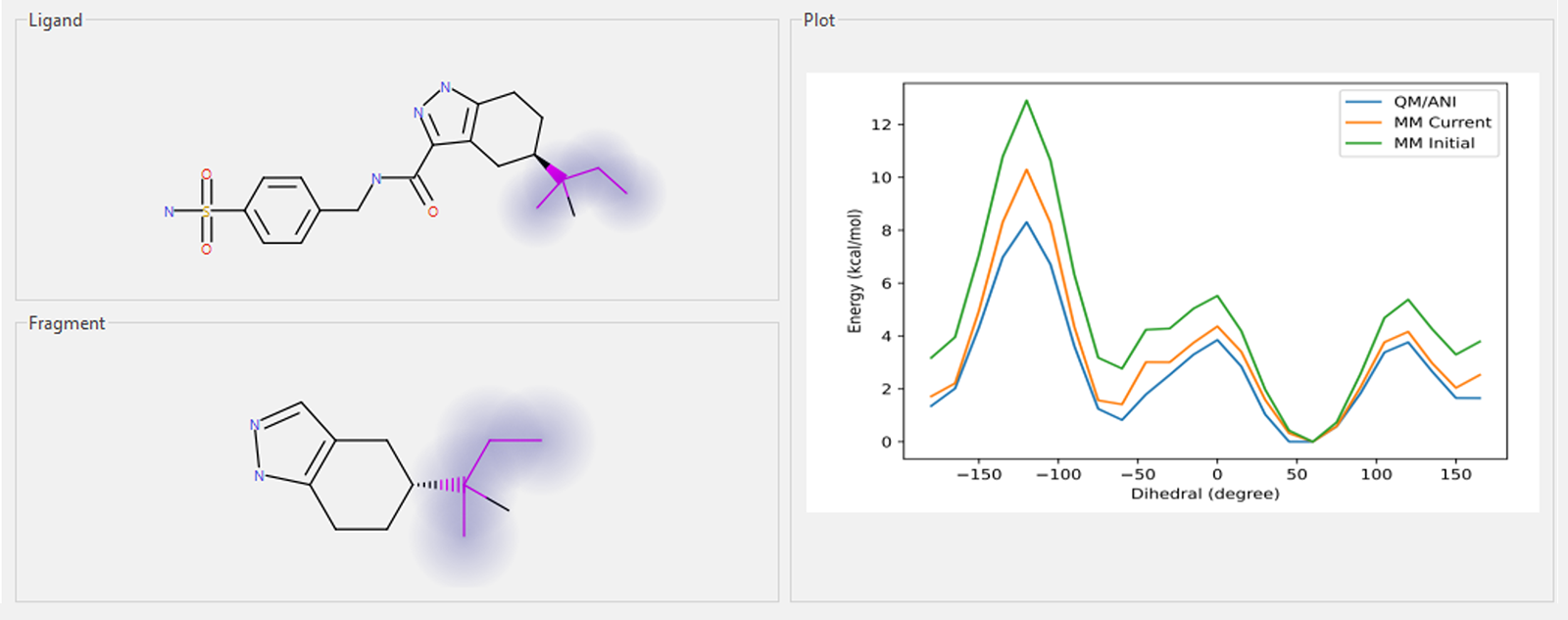
图2. 某些配体的二面角在所选力场下的描述不够准确。通常可以通过量子力学(QM)计算对这些二面角的参数进行优化,使其表现出更合理的物理行为。
构建一套能够精确描述各类配体与其生物大分子环境之间相互作用的通用参数集,至关重要。回望五年前,Cresset 加入“开放力场倡议”(Open Force Field Initiative)——这是一个由学术界与工业界科学家共同组成的协作组织,旨在开发(最初)一种高精度的配体力场,可与 AMBER 等生物大分子力场协同使用。过去五年中,OpenFF 力场已取得多项重要改进,未来还将持续演进……
然而,这一进展仍带来一个关键问题:基于配体的力场与生物大分子力场之间如何相互作用(或是否能有效耦合)?近年来,对共价抑制剂在结合位点环境中进行建模的需求日益增长。但由于缺乏连接两类体系的参数,准确模拟共价体系仍然十分困难。目前,业界正持续推动在统一力场框架下更精确地描述配体与蛋白质之间的相互作用。我们也在积极探索未来可靠建模此类共价体系的新方法与策略。
电荷
在我的最初博客文章中,我提到在相对结合自由能(RBFE)研究中准确模拟电荷变化存在困难,并建议所有分子应保持相同的形式电荷。然而,这一要求并不总是可行的;若将带电配体从数据集中剔除,会损失重要的信息。如今,通过引入反离子来中和带电配体,我们得以在扰动图中实现(图3)。通过延长电荷变化扰动过程中各λ窗口的模拟时间,能够在相对结合自由能实验中可靠地模拟电荷变化。
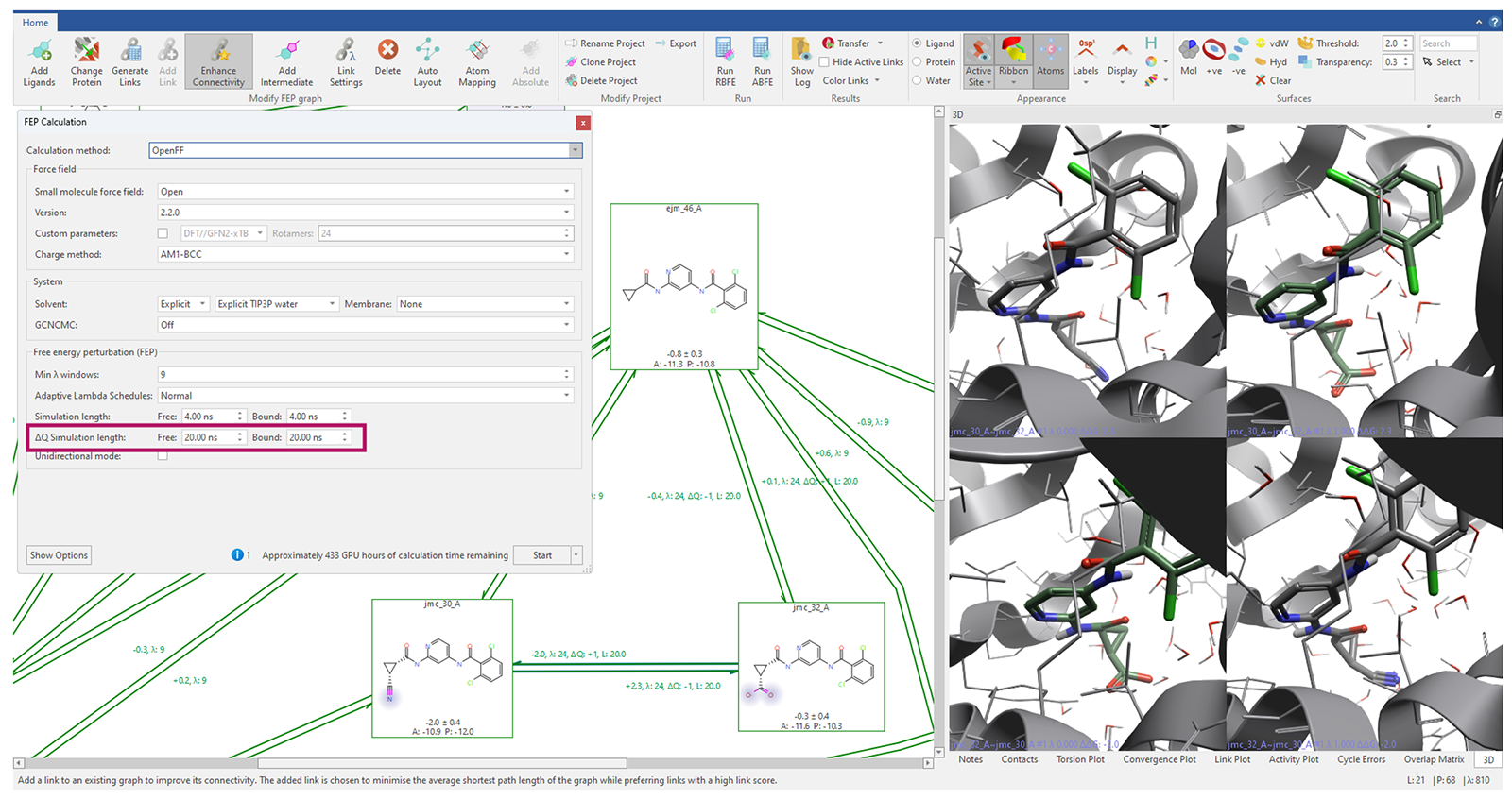
图3. 通过延长电荷变化转化过程中各λ窗口的模拟时间,能够在相对结合自由能实验中可靠地模拟电荷变化。
水分子
在分子模拟中,水分子的位置至关重要,这一点在FEP计算中尤为突出。RBFE计算容易受到不同水合环境的影响:如果某一连接路径正向转化中的配体与反向转化起始配体的水合环境不一致,则可能导致正向与反向转化之间的ΔΔG计算结果出现延滞现象(hysteresis)。因此,在FEP实验中确保所有配体都得到充分且一致的水合环境,是获得可靠结果的关键步骤。
可以借助3D-RISM和GIST等方法,分析体系中初始水合不足的位置,从而识别潜在问题区域。同时,采用如巨正则系综非平衡候选蒙特卡洛(GCNCMC)等采样技术——该方法通过蒙特卡洛步长实现水分子的动态添加与移除——能够有效提升配体周围水合环境的准确性,确保其在模拟过程中处于合理且物理上可信的水合状态。
靶标
可供研究的靶点种类繁多!一个显而易见的选择是聚焦于“简单”靶点——即水溶性蛋白,通常由数百个氨基酸组成。但面对更具挑战性的靶点又该如何?例如,位于膜上的靶点(如G蛋白偶联受体GPCRs)往往需要模拟数以万计的原子,对计算资源和处理器时间要求极高。然而,耐心是成功的关键——通过合理设置,完全可以在这些大型体系上开展模拟,并获得非常出色的结果。
在完成这些高成本计算、充分理解了不同条件下结果准确性的极限之后,现在我们便可以尝试对体系进行截断:即减少被建模的原子数量。这种方法有望显著缩短模拟时间,同时在很大程度上保持结果的质量,实现效率与精度之间的良好平衡。
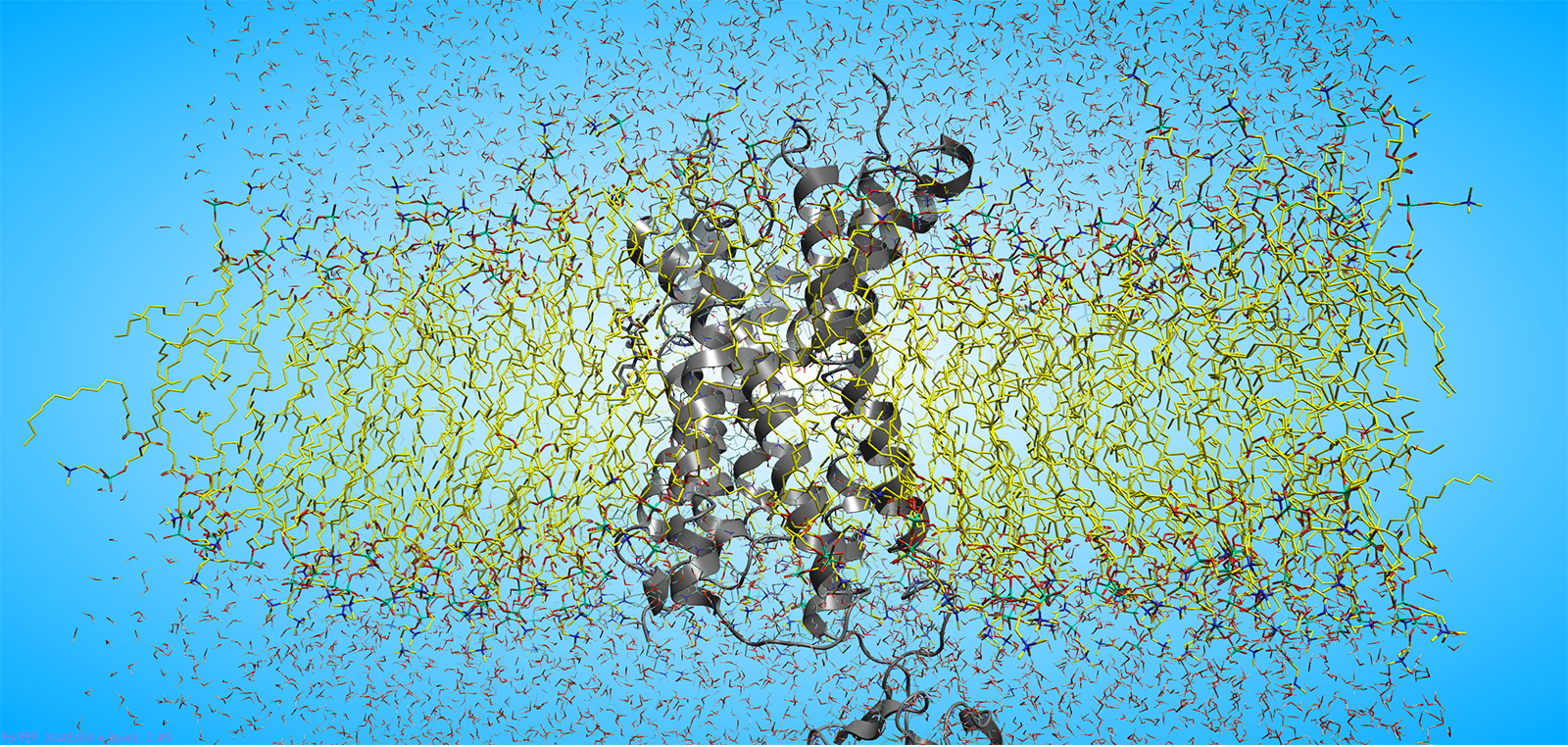
图4. 位于脂质膜中的蛋白质(黄色分子,如P2Y1受体,其二级结构以灰色卡通图表示)可借助相对结合自由能方法实现精确建模。
主动学习FEP
一种能进一步提升效率的工作流程是将FEP与3D-QSAR方法相结合:FEP模拟能够提供高精度的结合亲和力预测,但其计算过程固有地较为耗时;而更快速的QSAR方法则基于配体信息,可在牺牲一定准确性的前提下,迅速对大量分子集进行结果预测。当利用生物等排体替换策略(如Spark™软件提供的方法)或虚拟筛选研究(如使用Blaze™开展的工作)生成大规模的虚拟命中分子或设计分子集合时,可从中选取一部分分子进行FEP计算,再基于这些初始FEP结果,运用QSAR方法快速预测剩余分子的结合亲和力。
随后,将那些在初步筛选中表现出潜力的分子加入FEP计算集,并重新进行计算。这一过程不断迭代,直至结果不再出现显著改进为止。该主动学习策略有效融合了FEP的高精度与QSAR的高效性,实现了在保证预测质量的同时大幅降低整体计算成本。
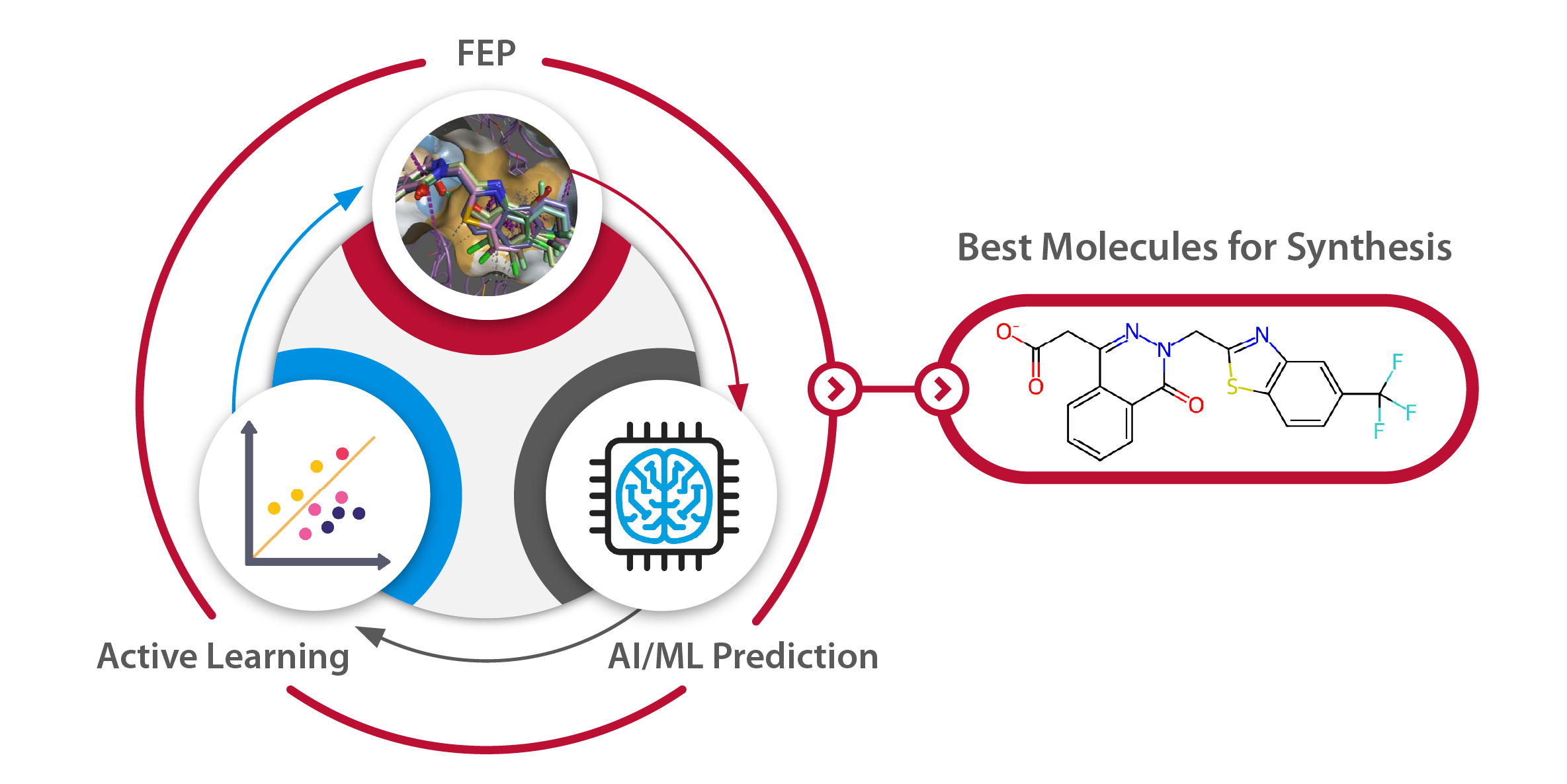
图5. 示意图展示了如何在一系列计算机设计分子中应用主动学习策略。
绝对结合自由能(Absolute FEP)
相对结合自由能(RBFE)的一个主要局限在于其可模拟的配体结构变化范围有限:通常情况下,一对分子之间的结构改变被限制在约10个原子以内。这一限制在药物研发的先导化合物优化阶段尚属合理——此时目标是优先排序已知类别的化合物——但在更早期的初筛阶段则显得极为受限,因为该阶段需要广泛探索化学空间(例如通过虚拟筛选实现)。而绝对结合自由能(ABFE)方法则提供了更大的灵活性:每个配体可独立计算,无需依赖其他分子作为参考。这种独立性使得ABFE能够处理更大范围的结构变化,从而更适用于早期药物发现中对全新化学结构的大规模探索与评估。
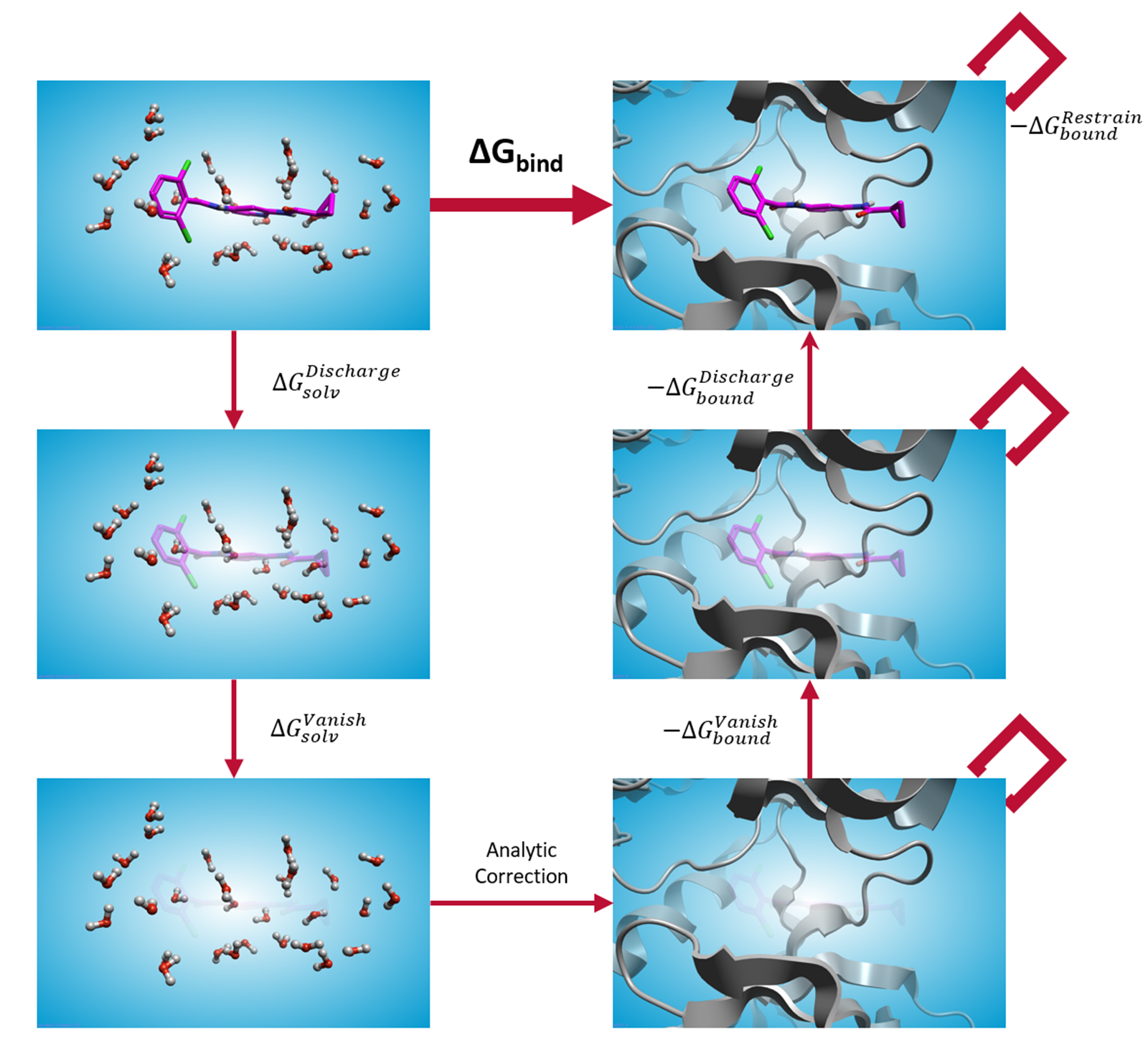
图6. 用于计算分子结合自由能的自由能循环示意图。在结合态和非结合态下,配体均通过两个步骤与环境解耦:首先关闭配体与环境之间的静电相互作用,随后再逐步关闭配体原子的范德华(vdW)参数。在结合态的计算过程中,配体始终被限制在结合位点内。
该方法的一个额外优势在于,无需对所有化合物使用完全相同的蛋白质结构。结合位点残基的质子化状态很可能受到所结合配体的影响。因此,在研究不同配体时,可根据其特性选用具有不同质子化状态的蛋白质结构,从而更真实地反映配体-蛋白相互作用的实际情况。
在绝对结合自由能(ABFE)计算中,与实验测得的结合自由能相比,常会出现系统性偏差(即“偏移误差”)。这通常源于对结合过程的简化描述——未充分考虑蛋白质本身在结合过程中发生的动态变化,例如残基质子化状态的改变。在标准的ABFE能量循环中,假设apo结合位点与holo结合位点具有相似的构象。然而,实际情况是:当配体接近蛋白质并开始相互作用时,蛋白质可能发生构象调整以适应配体。这种蛋白质构象变化(以及伴随的质子化状态调整)在大多数ABFE计算中并未被明确考虑,从而可能导致ΔG计算值与实验结果之间存在一定的误差。
运行ABFE计算比运行RBFE计算更为耗时,因为这类研究需要更长的平衡时间。例如,对一个同系物系列中的10个配体进行RBFE计算,可能仅需约100 GPU小时;而完成同等规模的ABFE实验,则可能需要高达1000 GPU小时。然而,这并不仅仅是GPU算力的问题:要充分发挥RBFE的潜力,通常需要科学家投入大量时间进行反复调试与测试,这对时间敏感的项目而言是极大的负担。
但随着我们自身Flare FEP平台适用范围的不断拓展,我们的科研团队对解决日益复杂的科学挑战积累了更深入的理解。这使得我们能够更有效地支持客户,为其项目提供最优的结果。
重新构想未来……
如果能克服当前部分模拟中的技术难题,ABFE将展现出巨大的潜力。以一种理想化的视角来看,它有望显著提升基于科学的产业效率。一个极具前景的应用场景是:在虚拟筛选实验中实现对初筛命中化合物的可靠选择。这类方法通常会探索广阔的化学空间,通过采购或合成一系列结构多样性的化合物并进行测试,以识别潜在的活性分子。随后,研究者通常会围绕这些已验证的命中分子进一步探索构效关系(SAR)。
与其依赖昂贵且耗时的“采购—测试—分析”循环,我们完全可以设想:利用ABFE技术首先精准识别出初始的多样化命中分子,再结合主动学习等策略,高效探索这些命中分子周围的邻近化学空间。这一流程将快速生成一个经过优化的候选化合物清单,明确指向最有可能成功、最适合目标靶点的合成分子。
因此,自由能微扰(FEP)方法具备释放数字化转型巨大潜能的潜力——通过在计算机中系统性地探索化学空间,精准识别出真正对项目关键的候选分子,以最高效的方式推动药物发现进程。
作者简介

Stuart Firth-Clark PhD
Stuart Firth-Clark在巴斯大学获得化学博士学位,其研究聚焦于利用计算方法探究化学反应性。此后,他在多家小型生物技术合同研究机构工作了20年,负责开发并实施面向潜在及现有客户项目的计算化学服务。2019年,Stuart Firth-Clark加入Cresset公司,担任高级应用科学家(Principal Application Scientist)。他主要负责管理Cresset的应用科学团队,并为公司客户提供培训与技术支持,帮助他们充分利用Cresset基于配体和基于结构的软件解决方案,实现各自的科研目标。
联系我们
Flare V10是Flare最新版本,交付先进的科学方法、分析工具和直观、易用的增强功能,洞察您的配体 – 蛋白质复合物结构。
想要尝试Flare信息丰富、用户友好界面,发现它如何帮助您自信地推动潜在先导化合物优化?请现在就联系我们安排试用,快速访问Flare的广泛功能。我们的专业团队随时准备通过安装和设置为您提供支持,而我们全面的教程库——涵盖从常见工作流程到高级方法和功能的所有内容将帮助您开始使用。我们在这里帮助您更快地实现目标,让您设计出感兴趣的分子。
电邮:info@molcalx.com
电话:020 – 38261356


