
Flare™ V11 正式发布:新增生成式 AI 工具、约束的蛋白质-蛋白质对接功能、分离计算模式以及众多其他增强功能
摘要:Flare V11 发布,显著提升计算药物发现效率与精度。新增 MolGenAI 基于 REINVENT4 和迁移学习,可生成符合目标理化性质的新分子;AI 助手提供对话与代码生成支持;蛋白-蛋白对接引入空间约束,助力Protac三元复合物建模;支持分离式计算,实现 HPC 任务断连运行;新增化学片段出现频次打分(Prevalence Score)评估分子可合成性;FEP 支持快速电荷计算、肽类分子与结果概览;分子动力学模拟支持 ZAFF 力场与多种轨迹格式文件导出;并优化大项目性能与配体嵌套角色管理。整体强化了从分子生成到分析的全流程能力。
原文:Sofia Bariami. December 9, 2025. Flare™ V11 released
我们非常高兴地宣布最新版 Flare V11 正式发布!此版本带来了一系列新增功能和性能增强,旨在让计算药物发现更加高效、精准且易于使用。从全新生成式化学工具、改进的自由能微扰(FEP)工作流、增强的蛋白-蛋白对接能力,到更智能的配体管理以及更灵活的计算处理方式,本次更新直面研究人员在处理复杂分子体系时所面临的关键挑战。
使用 MolGenAI 生成全新分子
MolGenAI 是本次版本中最令人兴奋的新增功能之一:这是一款基于先进人工智能技术的强大分子结构生成工具。它基于 REINVENT4 技术,使用在高质量数据集(如 ChEMBL)上训练的先验模型,使研究人员能够生成探索化学空间全新区域的分子。
MolGenAI 还支持迁移学习(transfer learning),允许用户使用少量示例化合物将现有先验模型重新聚焦到更窄的、用户自定义的化学子空间。这意味着您可以将分子生成过程量身定制,专注于与当前项目最相关的全新化合物。新训练出的先验模型可被保存、重复使用,并轻松在团队内部共享。
为进一步简化最优分子的筛选流程,MolGenAI 与 Flare 的 Radial Plot 打分无缝集成,可根据生成分子与目标理化性质的匹配程度进行过滤。通过调整整体或各单项打分阈值,用户可以灵活控制筛选条件,获得满足特定设计标准的分子。
例如下图所示,我们演示了如何生成一个已知配体的新类似物。本例中,我们以蛋白 PDB:1OIT 的共结晶配体为起点:
- 首先,我们构建了一组结构相关的化合物。此处我们使用了在Flare 中 Spark™ 的 R 基团替换功能,以确保保留关键骨架。
- 然后,以 Spark 生成的分子作为训练集,对先验模型进行再训练,使其偏向于目标化学空间。
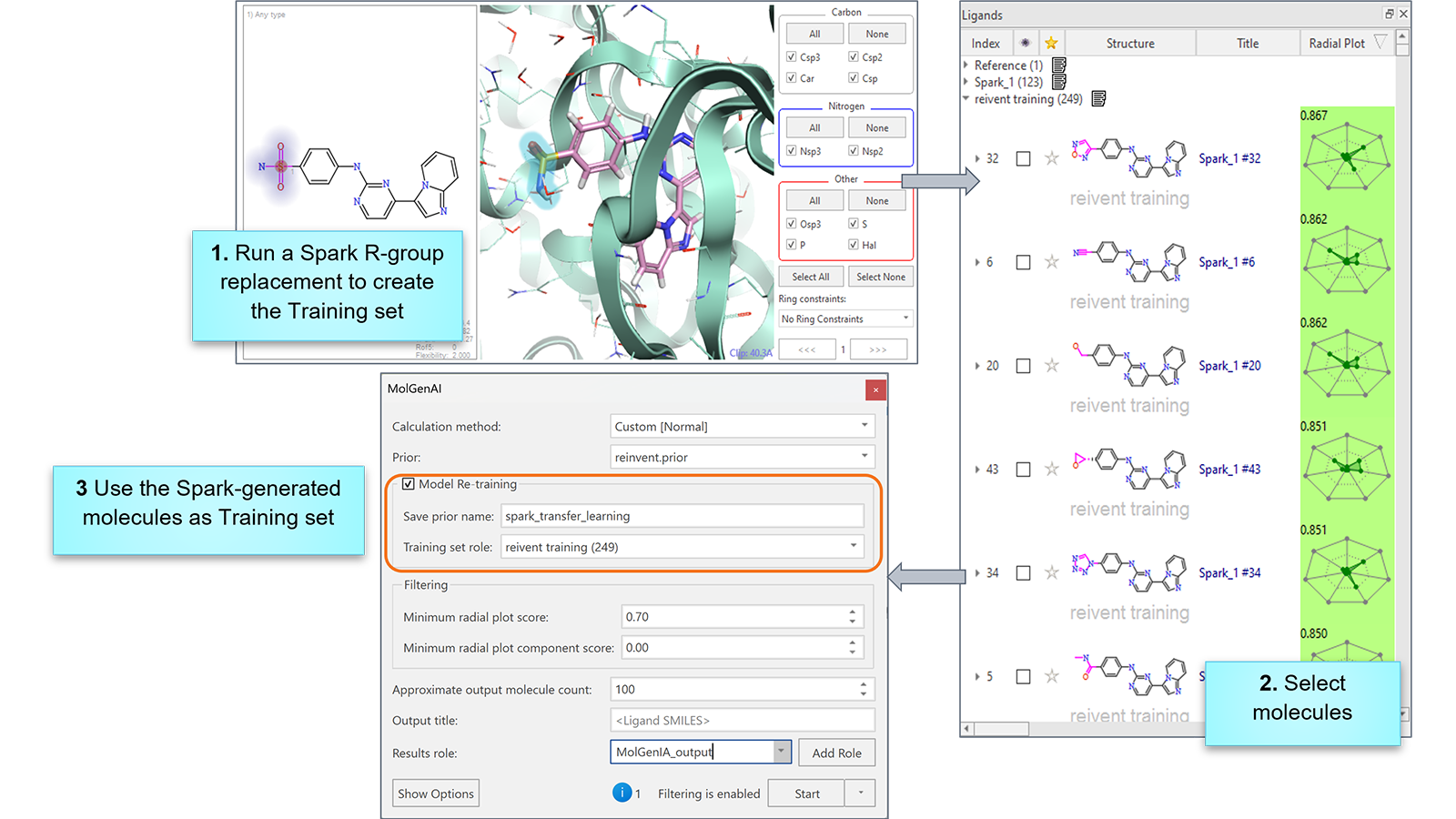
图1. 在 Flare V11 中,MolGenAI 能够生成探索全新化学空间的分子。现有先验模型可被重新聚焦于用户定义的更窄化学空间区域——例如,此处使用 Flare 中 Spark 生成的分子作为训练集,对模型进行再训练,使其偏向于目标化学空间。
过滤功能直接集成在 MolGenAI 工作流中:用户可通过“径向图属性(Radial Plot Properties)”控件设置关键性质(如分子量 MW、SlogP 和柔性)的范围/函数及权重。通过将每个打分成分的最低分值设为 0.7,即可仅保留与设计性质特征高度匹配的分子。
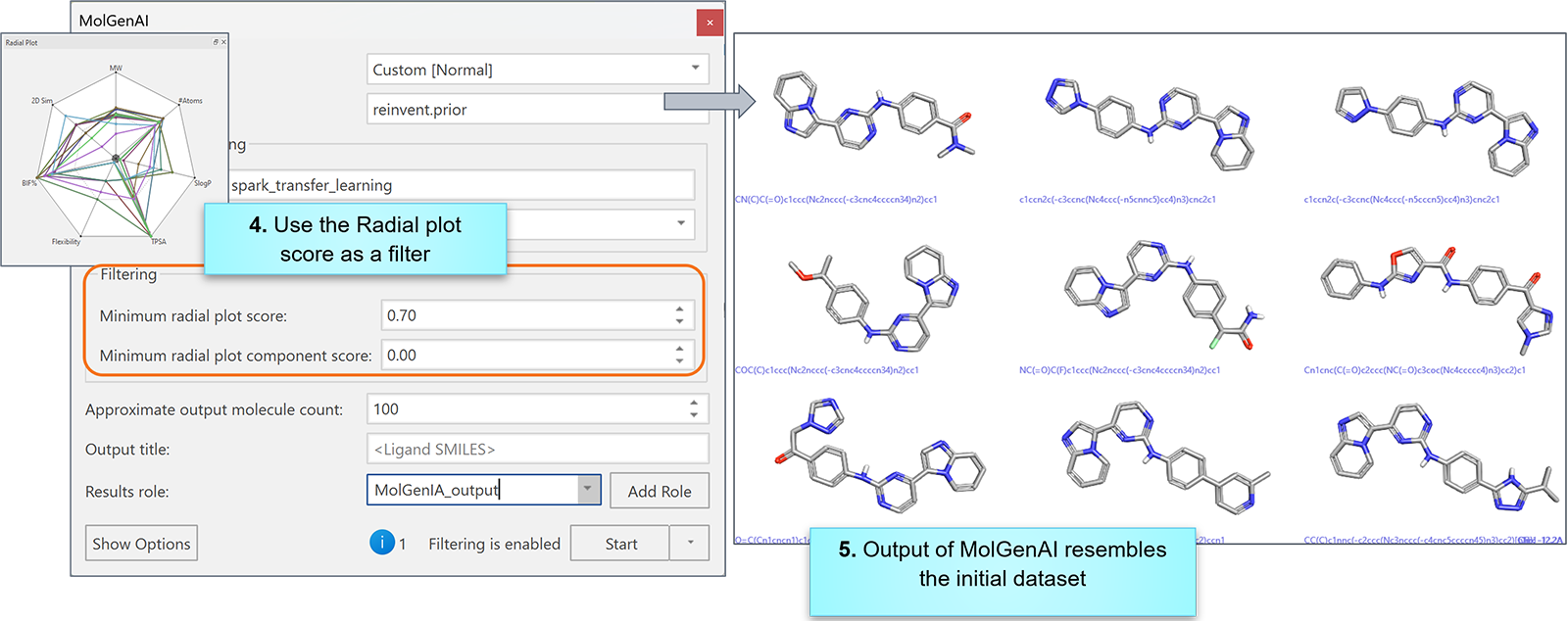
图2. 只需几个简单步骤,即可生成符合理化性质设计要求的全新分子构想。
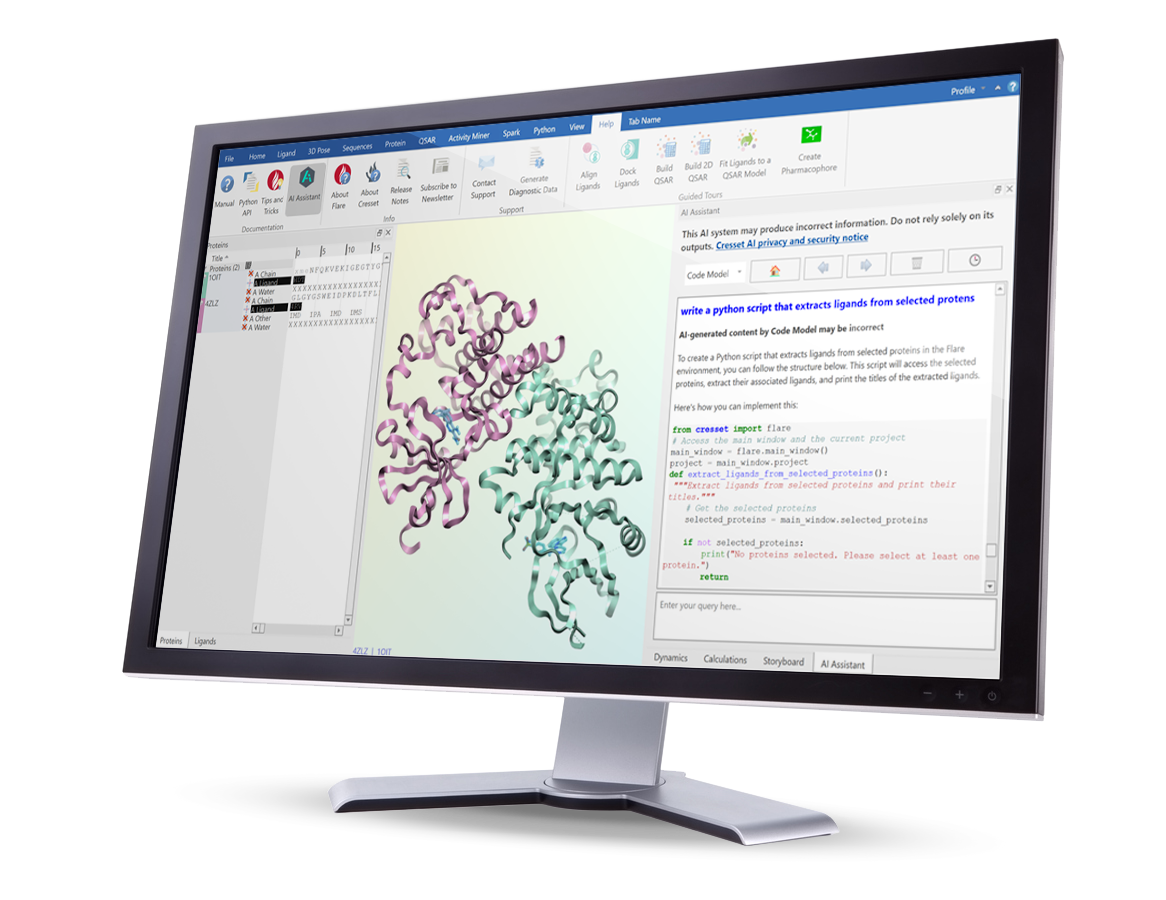
借助 Flare AI 助手,更智能、更高效地工作
本版本另一项新功能是 AI 助手(AI Assistant),旨在让 Flare 的使用更快、更轻松。助手包含聊天(Chat)模型和代码(Code)模型。Chat 模型可回答有关 Flare 通用操作及底层科学方法的问题——无论您是想快速回顾某个实验如何设置,还是需要了解某项计算背后的理论原理,Chat 模型都能在您的工作环境中直接提供指导。
Code 模型则担任“编程副驾”(copilot),为使用 Flare Python API 的用户提供强大支持。它能帮助您快速编写和自定义脚本,将想法迅速转化为可执行代码。脚本创建后可直接在 Flare 中运行,轻松扩展功能或实现常规任务自动化。
AI 助手的两大模型共同作用,不仅让新用户更易上手 Flare,也赋能高级用户更快工作并共享其工作流。
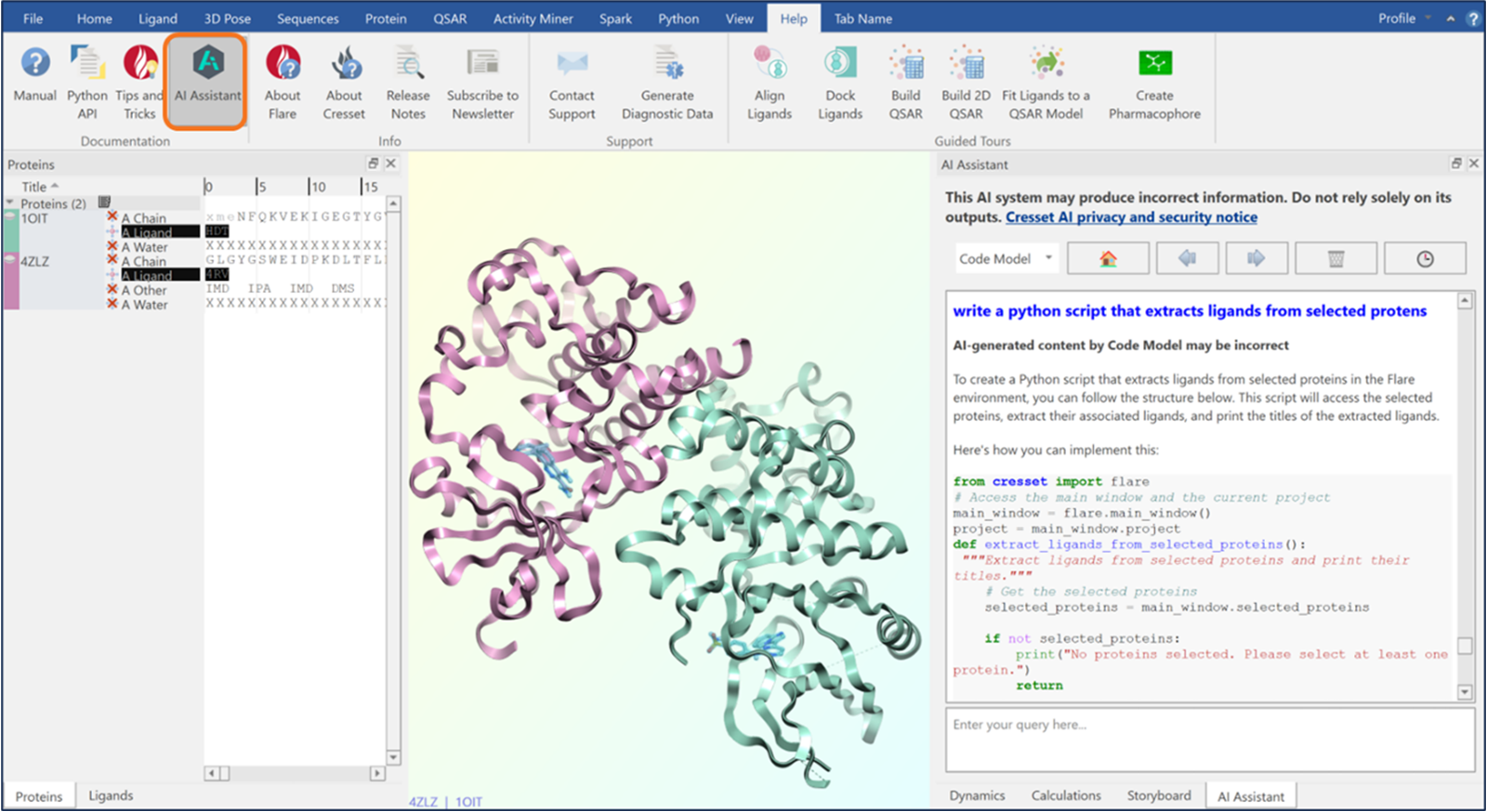
图3. AI 助手的 Code 模型可生成脚本,帮助简化 Flare 中的各项任务。
带约束的蛋白-蛋白对接
在蛋白-蛋白对接(PPD)实验中引入约束条件,是对这一热门方法的重要增强。通过策略性地施加约束,现在可在对接两个蛋白链的同时,引导它们之间的空间取向。这些约束确保蛋白间的相对取向既物理合理,又符合预期的结合构型,为生成三元复合物的起始结构提供了一种高效方法。
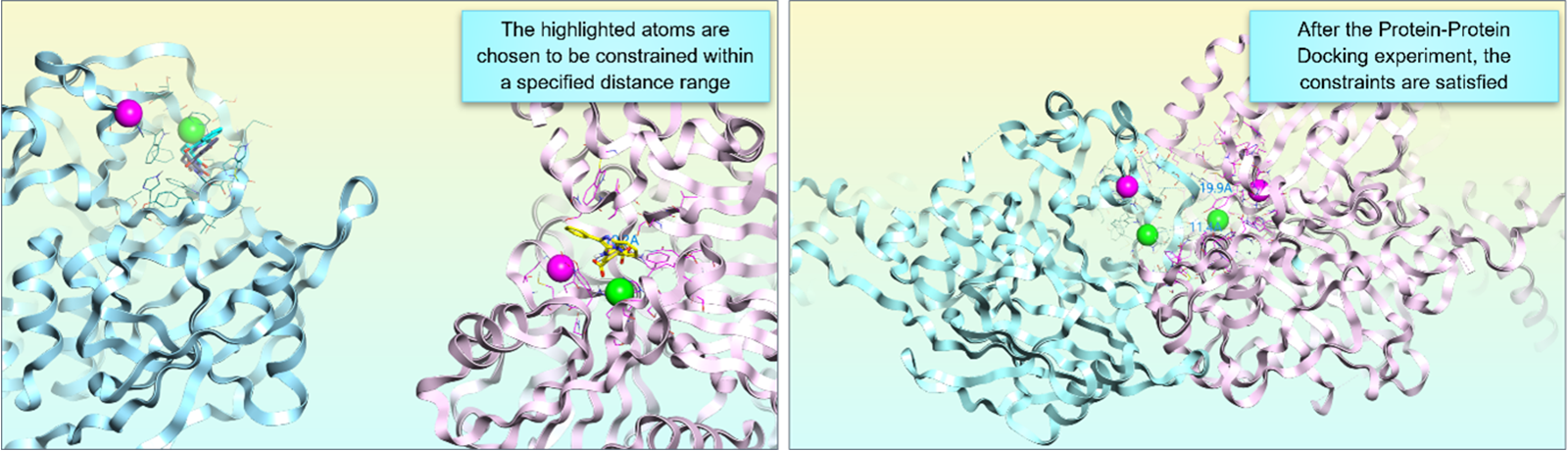
图4. 在同色原子之间施加 5–20 Å 范围的约束。蛋白-蛋白对接完成后,约束条件得到满足:粉色原子间距为 20 Å,绿色原子间距为 11 Å。
靶向蛋白降解剂(TPD)的设计便是约束性蛋白-蛋白对接的关键应用场景。在受控条件下完成蛋白对接后,该体系可作为后续研究的初始支架——蛋白正确取向且结合位点保持可及,便于在后续建模步骤中引入配体或连接臂分子。
分离式计算(Detached Calculations)
Flare V11 与 Cresset Engine Broker(CEB)V3.5 联动,引入了全新的“分离式计算”功能,为管理计算密集型任务提供了高效方式。此前,通过 GUI 提交的长时间计算任务需保持 Flare 界面运行,限制了灵活性。现在,任务一旦启动,Flare 会将所有输入数据传输至 CEB,随后即可安全断开连接——用户可自由关闭 Flare 而不影响工作流。
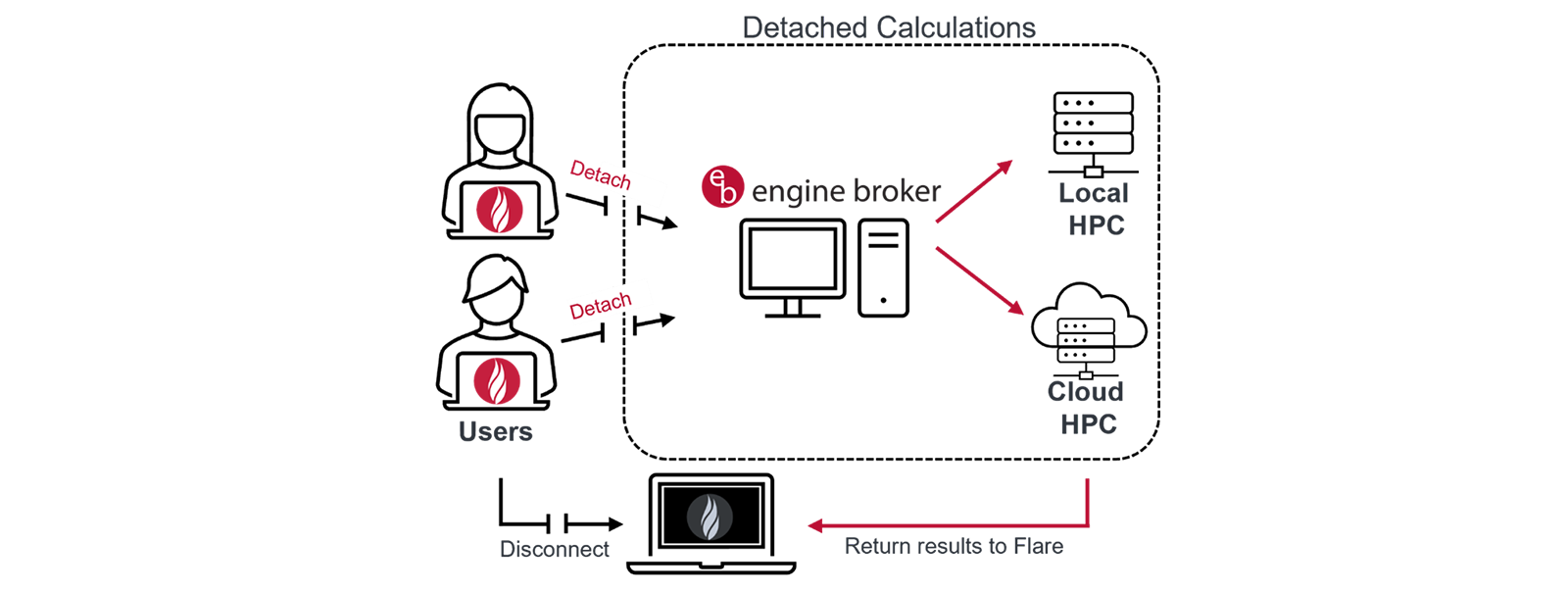
图5. 分离式计算允许用户完全断开 Flare,而计算任务继续在 HPC 集群上运行。此功能需搭配 Flare V11 与 CEB V3.5。
后台,CEB 会自动启动一个专用后台进程独立处理计算。进度与错误信息将在 Flare 专用窗口中持续更新。计算完成后,结果将存于后端,并可立即下载并整合到当前项目中。
该新架构带来多重优势:降低因意外关闭或系统重启导致工作丢失的风险;通过将 GUI 与计算解耦,减少本地资源占用;并支持对大型或耗时任务更灵活地调度计算资源。
出现频率打分(Prevalence score)
Flare V11 新增了出现频率评分(Prevalence Score),帮助用户快速评估目标分子片段的常见程度,从而关联其可合成性。该方法基于 Spark 数据库(ChEMBL、eMolecules Commercial Screening 和 eMolecules Reagents)中精心整理的片段频率数据,提供一种简单、定量的合成可及性估计。
出现频率评分通过将分子片段化,并评估这些片段在上述数据库(即实际可合成化合物中)出现的频率来计算:常见且易得的片段打分接近 1,稀有或结构复杂的片段则接近 0。各片段打分可在 Flare 中可视化,而整个分子的出现频率打分为其所有片段中的最低分值。该分数范围为 0(极低出现频率/极复杂)至 1(高出现频率/极简单),并显示在配体表的“Prevalence Score”列中。
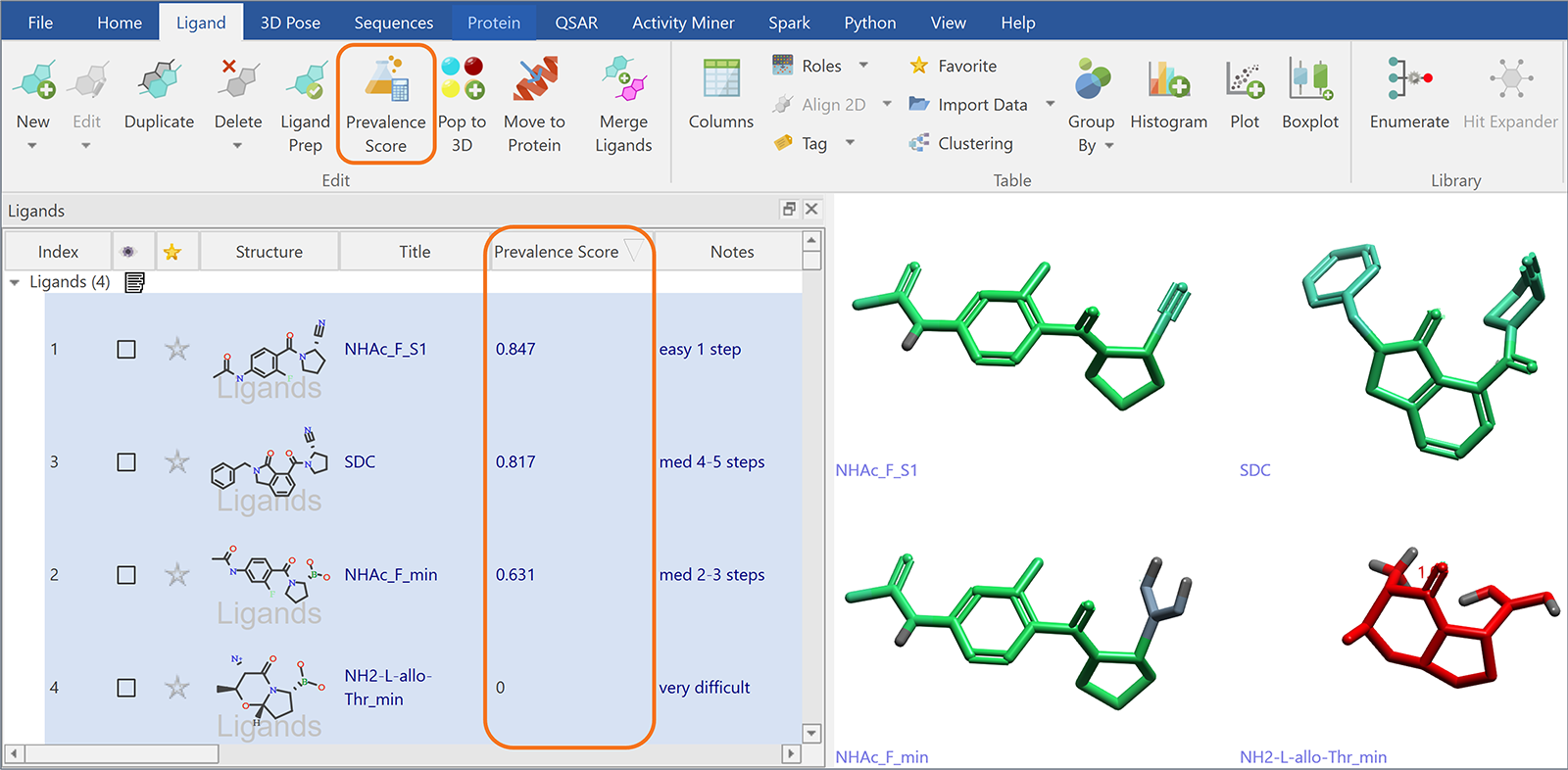
图6. 配体表单中的“出现频率打分(Prevalence Score)”列告知用户各分子所含片段的常见程度,并可关联其可合成性。整体分子打分由最不常见的片段决定,范围从 0(极低出现频率/极复杂)到 1(高出现频率/极简单)。
为便于解读,Flare 还提供“按出现频率打分着色”的选项,高亮每个原子/片段对打分的贡献。配体从绿色(常见)到红色(罕见)着色,用户可一眼识别可能阻碍合成的罕见结构区域。这种交互式视图便于快速定位分子中的问题部分,指导设计朝着更易合成的化合物方向优化。
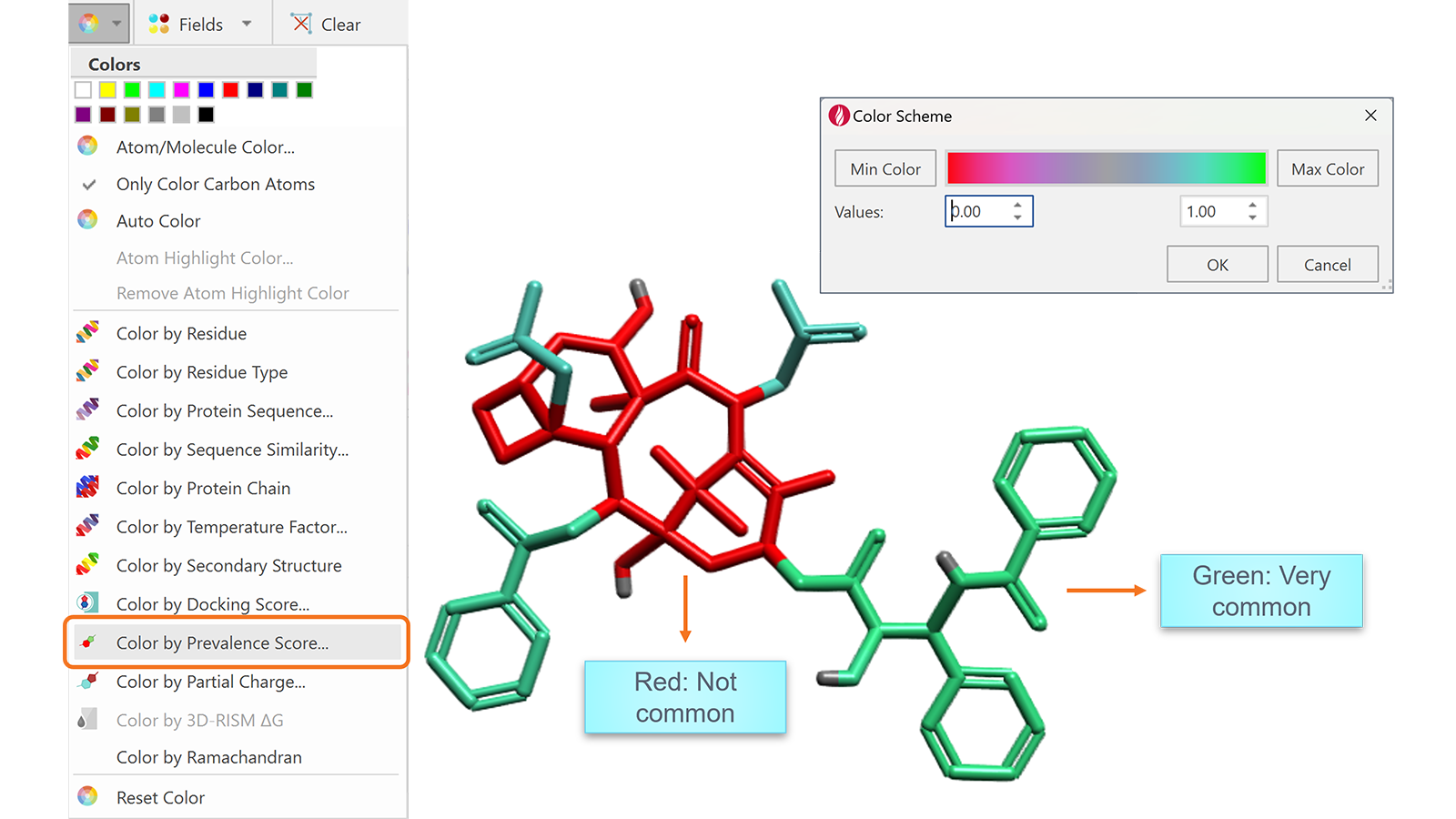
图7. 整体分子的出现频率打分为最不常见片段的最低分。本例中,打分为 0,因为分子中心部分(红色所示)结构极其复杂,在片段数据库中极为罕见。
性能提升与嵌套角色(Nested Roles)
Flare V11 已全面重构,可处理规模大得多的项目。处理数万个配体和数百个蛋白的研究人员将发现 Flare 响应更迅速、运行更稳定。得益于此,内存占用显著降低,使用户能更轻松处理大型数据集而不受系统限制。
本版本另一亮点是“嵌套配体角色”功能。它在配体表单中引入层级结构,允许用户为分子分配“父级”和“子级”角色。这在单个化学实体存在多种变体(如不同质子化态、互变异构体或对接构象)时尤为有用。创建嵌套角色非常简单:在设置新角色时选择父级,或在 Python API 中使用斜杠语法即可。这个特性可灵活地帮助研究人员更好组织数据,在复杂化学关系中进行高效分析。
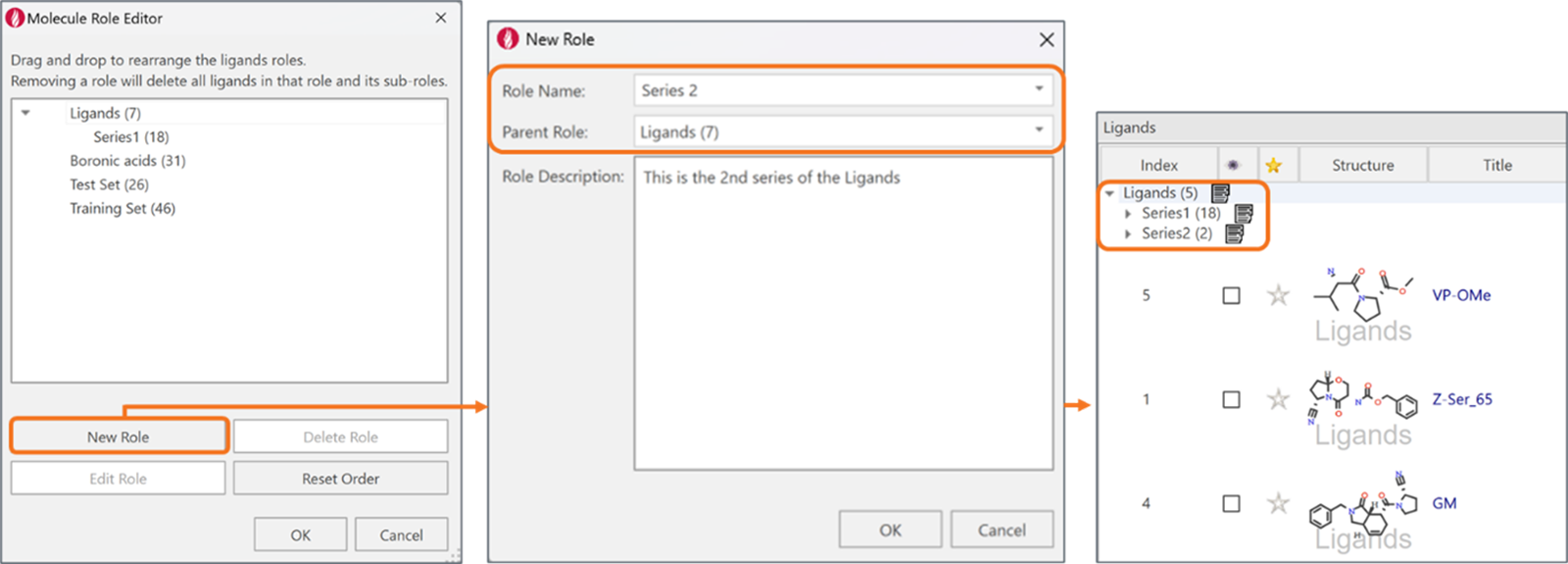
图8. 使用嵌套角色改进配体表单中分子的组织方式
Flare FEP 改进:支持快速的肽类计算
Flare V11 中的 FEP 功能进行了多项优化,旨在提升速度与易用性。
关键改进之一是:通过 AshGC(NaGL)机器学习方法,为配体计算 OpenFF 偏电荷。该新方法在保持精度的同时实现极快的电荷分配,使肽类等大型复杂分子的参数化变得切实可行,并顺利纳入 FEP 研究。此增强也适用于所有包含小分子参数化的计算,如能量最小化、分子动力学和 GIST。
FEP 图的生成速度也得到大幅提升,尤其在处理大分子和肽类体系时,用户可比以往更高效地设置并执行肽类 FEP 计算。
为支持分析,Flare FEP 项目新增了“Overview”(概览)标签页(见图9)。该标签页将 FEP 结果集中展示,同时显示预测与实测 ΔG 值(如有)及配体结构信息,便于用户快速评估结果质量,并轻松导出与团队共享。
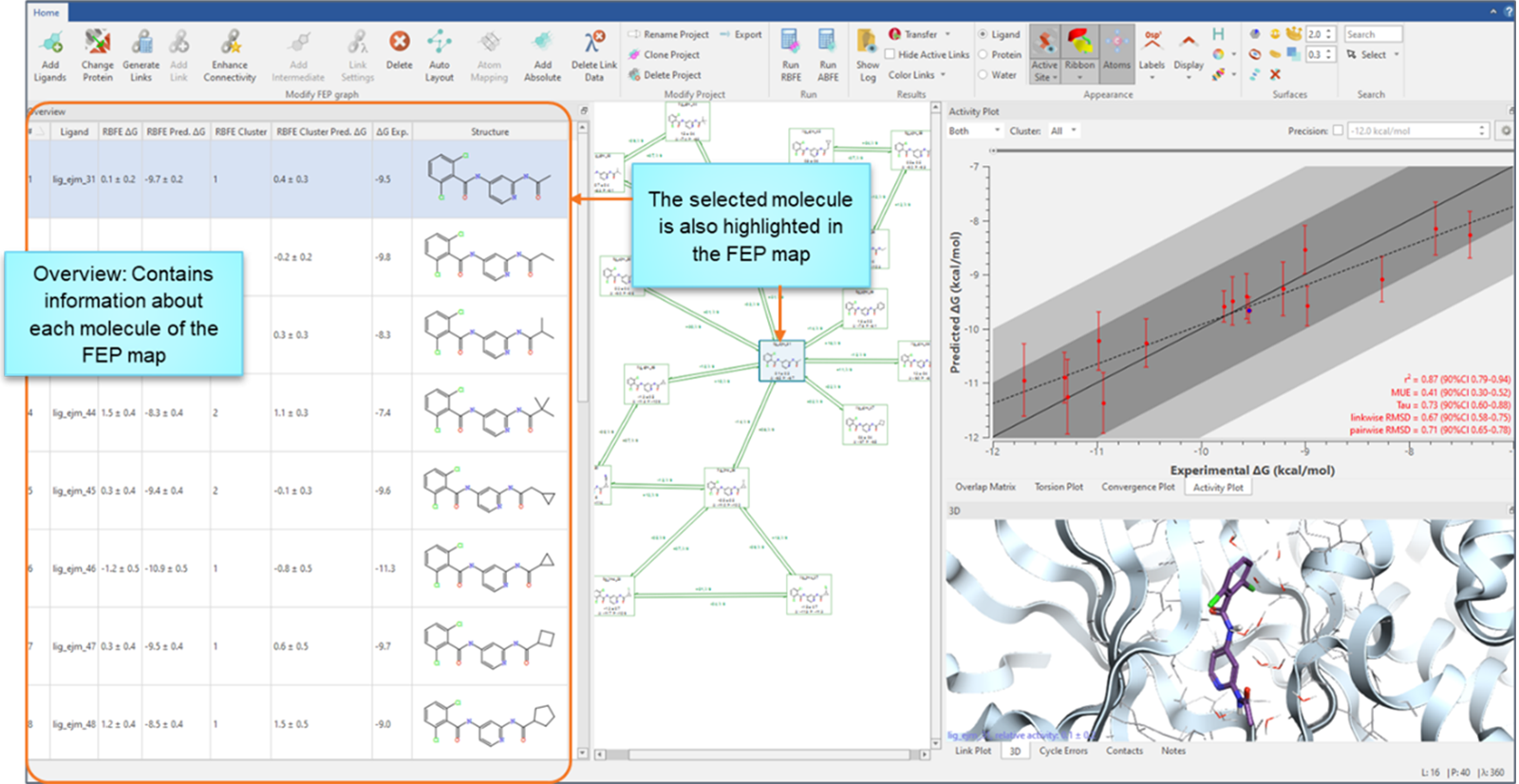
图9. FEP 结果概览标签页可与 FEP 图及 3D 视图交互
对于绝对 FEP 计算,Boresch 约束参数及约束强度算法已根据更广泛的科学验证结果进行了精细调整。约束定义现在更准确地反映蛋白-配体间的典型成键与非成键相互作用。此外,当仅运行绝对 FEP 计算时,系统会计算相对于实验值的偏移量,并直接应用于活性图中,从而更清晰地展现预测性能。
图10展示了Flare V10 与 V11的活性图比较结果,在V11的活性图中直接应用了针对绝对自由能微扰计算相对于实验值的计算偏移量。
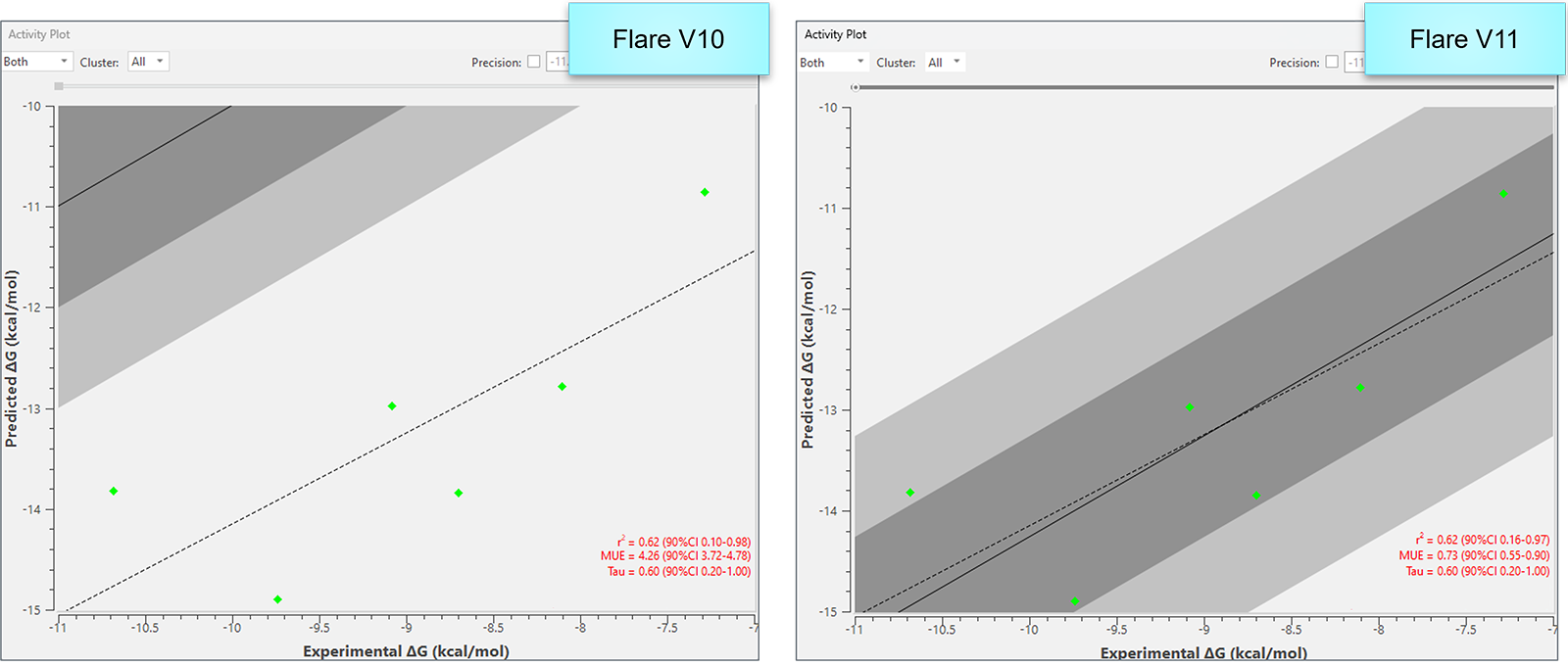
图10. 在绝对自由能微扰基准研究中,计算并应用了一个偏移量至活性图数据点,从而更清晰地展示模型的性能表现。
分子动力学(Dynamics)的新增功能与改进
分子动力学计算引入了多项关键改进,既拓展了支持体系的范围,也增强了轨迹分析工具。
AMBER 力场现已升级,支持 ZAFF 锌力场,可更准确地模拟金属蛋白及含锌复合物。轨迹还可导出为 XTC 格式(见图10),提供一种广泛兼容且存储高效的新选项。
工作流性能也得到优化:动力学模拟过程中 RMSD 图的自动更新已被禁用,以避免不必要开销。用户现可按需手动刷新图表,从而更好地控制计算资源。
分析与可视化功能亦有提升:主成分分析(PCA)现在可基于用户自定义的帧子集进行,而非局限于整个轨迹,使研究人员能更精细地控制构象采样范围。轨迹动画查看器新增了播放速度控制,便于在不同的时尺度检查运动,聚焦关键结构事件。
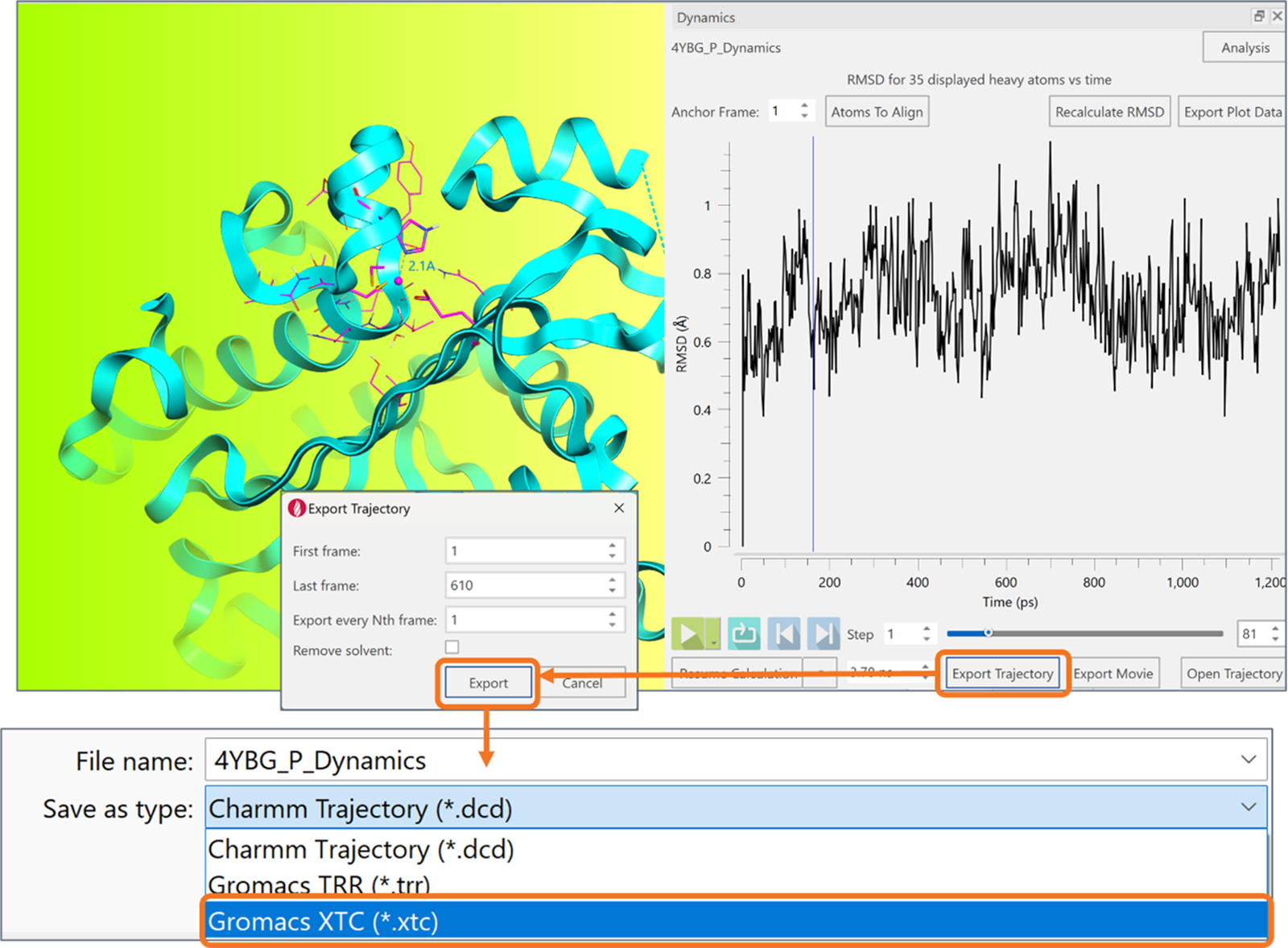
图11. Flare V11 支持将分子动力学轨迹导出为除 dcd 之外的多种文件格式。
Flare V11 其他亮点
每个 Flare 版本的开发都围绕两大优先级:将最新科学进展引入平台,并让日常使用更流畅、更直观。许多改进源于用户社区的反馈。以下是 Flare V11 中更多值得关注的新功能与增强:
- 所有 Flare 计算均支持 5 位字母的残基名称
- 编辑菜单中新增椅式和扭船式六元环构象模板
- Flare 的“仅打分”计算及 Spark 对接计算中启用“闪烁水分子”(flickering waters)
- 新增库枚举反应:包括偕二醇转化为酮/醛、7-氧杂双环[2.2.1]庚烷芳构化等
- 新增选项:将 Activity Miner 的 Top Pairs 表导出为 SDF 格式
用 Flare V11 加速您的研究
Flare V11交付先进的科学方法、分析工具和直观、易用的增强功能,洞察您的配体-蛋白质复合物结构。
想要尝试Flare信息丰富、用户友好界面,发现它如何帮助您自信地推动潜在先导化合物优化?请现在就联系我们安排试用,体验Flare的最新功能。我们的专业团队随时准备通过安装和设置为您提供支持,同时我们丰富的教程库(从基础工作流到高级方法)确保您顺利上手。借助 Flare V11,您将能够更快推进研究、深入洞察机制,并设计出最具价值的分子。
- 电邮:info@molcalx.com
- 电话:020-38261356


