
摘要:水分子在蛋白–配体相互作用中扮演着关键但常被忽视的角色,其中高能水(即热力学不稳定的结合位点水)的置换可显著提升配体结合亲和力,已成为基于结构的药物设计(SBDD)中的重要设计策略。本文以 15-PGDH 抑制剂的先导化合物优化为案例,系统复现并深入分析了 Dodda 等人通过水引导设计实现活性提升逾 400 倍的研究工作。借助 Flare 平台实现的 GIST 方法,对 15-PGDH–化合物 1 共晶结构(PDB 9PFL)结合口袋进行了 apo-GIST 分析,识别出子口袋中多个热力学不利的水合位点。结果表明,先导化合物 4 仅实现对高能水的部分置换,而优化后的化合物 38 则完全取代两个关键高能水分子,从而获得显著的结合自由能增益。在该优化路径中,传统对接打分函数(如 Lead Finder 的 LF-dG 与 GNINA 的 Affinity)以及 FEP+ 方法均未能充分反映由水置换主导的自由能变化(预测 \(\Delta \Delta G \approx 0.7\text{–}1.4\ \text{kcal/mol}\),实验值为 \(3.62\ \text{kcal/mol}\)),反映出在未显式建模水的前提下,其对该类效应的刻画能力存在局限。相比之下,在保持原有对接框架不变的情况下,引入基于 GIST 的配体水置换自由能项(\(\Delta G_{\text{watdisp}}\))作为独立修正层,对打分结果进行补偿,可在该体系中合理再现实验观测的 ΔΔG(预测 4.40 kcal/mol vs. 实验 3.62 kcal/mol)。本研究表明,GIST 不仅可用于识别具有设计价值的高能水合位点,更可作为一种显式引入溶剂热力学信息的物理补充层,与现有对接与自由能计算方法形成互补,为以水为中心的先导化合物优化提供了具有可操作性的计算策略。
肖高铿/2026-01-16
前言
15-羟基前列腺素脱氢酶(15-PGDH)作为调控前列腺素 E2(PGE2)代谢稳态的关键限速酶,通过催化 PGE2 氧化失活参与调控其生物活性。鉴于 PGE2 在免疫调节、炎症响应及组织再生过程中的核心调控作用,靶向抑制 15-PGDH 活性已成为开发组织再生增强型治疗策略的重要研究方向。
Dodda 等人1基于 15-PGDH 与抑制剂化合物 1 的共晶结构解析结果,将化合物 1 的分子骨架划分为三个可修饰区域(图 1),并针对各区域开展系统性结构优化研究,并实现了先导化合物优化。
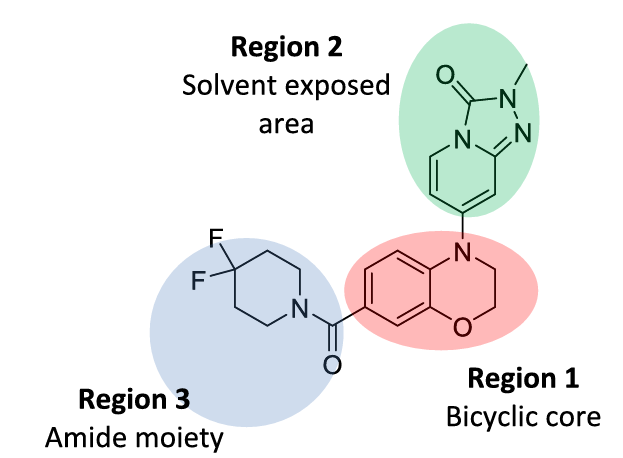
图1. 基于共晶结构将抑制剂1分为三个可修饰区域,分别进行结构优化
其中,针对 Region 2 (三唑并吡啶酮)的结构优化策略源于 WaterMap 水合自由能分析所揭示的关键信息:15-PGDH 与化合物 1 的结合口袋中存在具有高去溶剂化能的高能水分子,通过分子设计引入特定官能团取代该类高能水分子,可通过降低结合自由能显著提升抑制剂与靶点的结合亲和力。研究结果表明,化合物 38(图 2)通过定向置换结合口袋中的高能水分子,其生物活性相较于先导化合物 4(图 2)实现了 400 余倍的显著提升,验证了该优化策略的有效性。
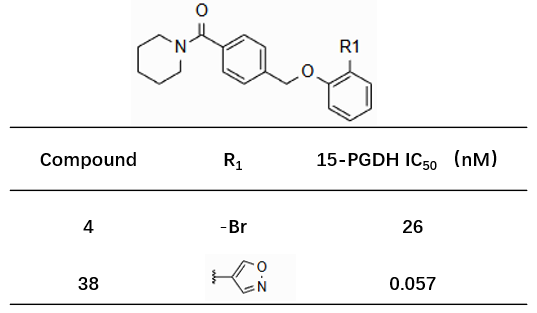
图2. 15-PGDH抑制剂4与38的化学结构对比
网格非均匀溶剂化理论(Grid Inhomogeneous Solvation Theory, GIST)2 是一种基于分子动力学模拟的三维网格化分析方法。该方法将溶剂(如水)的热力学性质(包括能量、熵及自由能)离散化至受体结合位点周围的高分辨率空间网格上,从而提供水合位点及其热力学性质的空间分布。在基于结构的药物设计(Structure-Based Drug Design, SBDD)中3,GIST 可用于识别配体结合过程中可能被置换的水分子区域,尤其是那些相对于体相水具有不利自由能(即热力学不利)的高能水位点(或称为unhappy water)。据此,可指导配体设计:一方面,将配体官能团延伸至这些高能水区域以实现水置换;另一方面,避免扰动能量上有利(即稳定的)水分子(或称为happy water),从而理性预测并优化配体的结合亲和力。更多的GIST在SBDD中的应用案例,参见前文4。
本文旨在借助药物设计平台 Flare5 实现的 GIST6 水分析技术,对 15-PGDH – 化合物 1 共晶复合物的结合口袋进行精准表征。通过复现 Dodda 等人1基于结合位点高能水分子发现先导化合物优化契机的研究思路,从定性与定量层面阐释化合物 4 到化合物 38 活性提升逾 400 余倍的机制,为基于水分子热力学特征的 SBDD 实践提供方法学参考与技术范例。
结果
15-PGDH结合口袋的apo-GIST分析:发现先导物优化的契机
首先用Flare分析化合物1与15-PGDH的共晶结构(PDB 9PFL),在化合物1的溶剂暴露区域(图1,Region 2)处,可以发现有个被水(HOH483、485、486)占据的结合子口袋,如图3所示,化合物1的三唑并吡啶酮的羰基氧那一侧朝向子口袋,并与 HOH483、485 及 486 等水分子相邻。
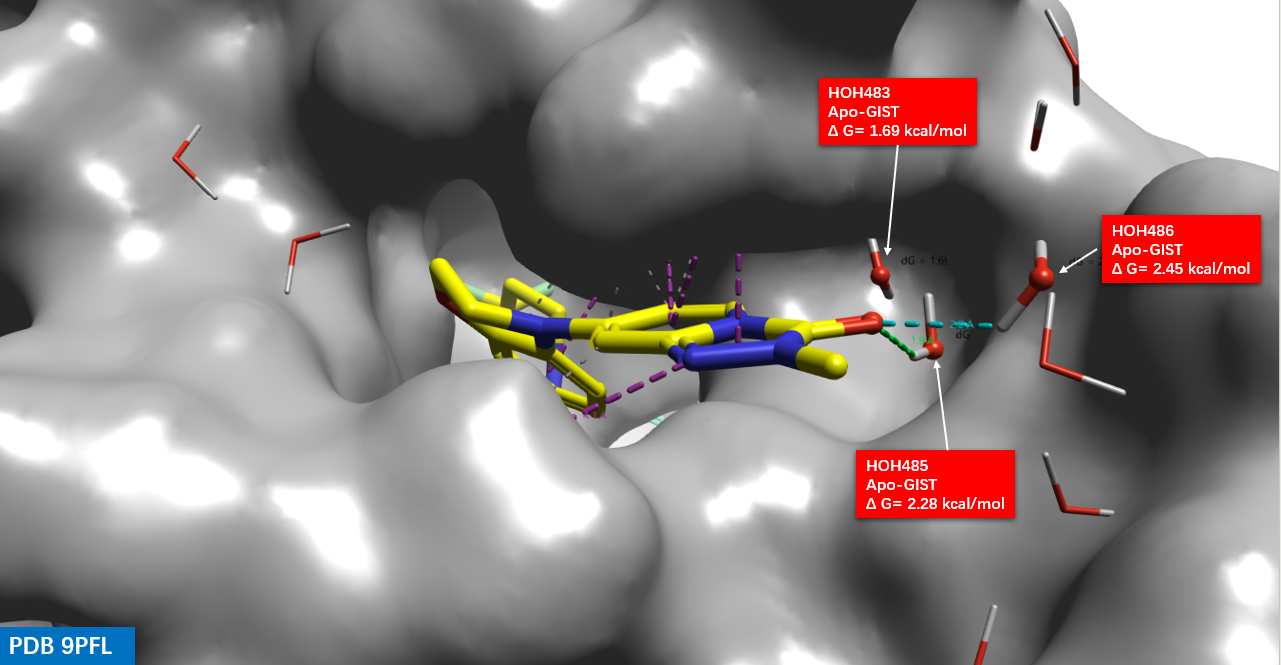
图3. 化合物1与15-PGDH共晶结构(PDB 9PFL)结合位点及其关键水。分子表面:15-PGDH,黄色棍棒:化合物1;细棍状:水。
基于 GIST 水分析指导先导化合物优化的物理原理在于:若配体中的重原子取代了结合口袋中具有高占据率且热力学不利(即”unhappy”)的水分子,则该配体将获得有利的结合自由能贡献。因此,有必要计算结合子口袋内水合位点的热力学性质,以判断其是否为”unhappy water”。

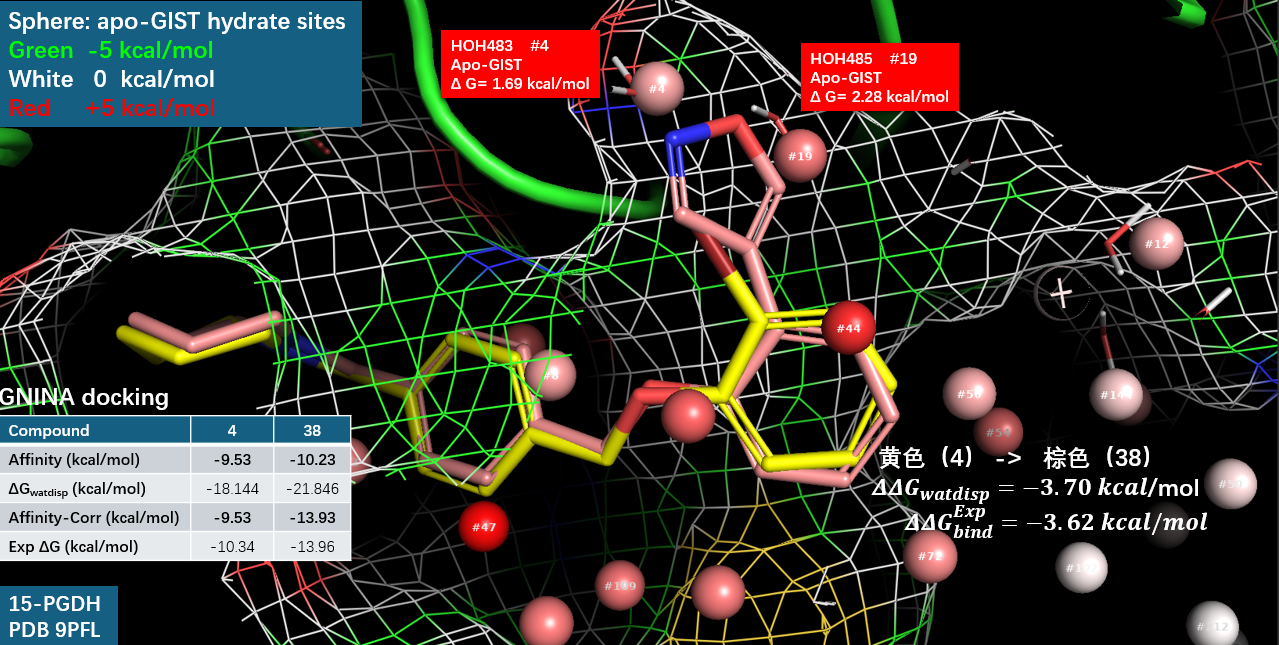
图4. 化合物1与15-PGDH共晶结构(PDB 9PFL)结合位点以及GIST预测的水合位点。分子表面:15-PGDH,黄色棍棒:化合物1;线状:共晶水;球状:GIST预测的水。
Flare GIST 实验的具体方法参见文献 4。在本研究中,对 PDB 9PFL 的结合口袋进行了 apo-GIST 分析,并基于水密度进行了水合位点预测。结果如图 4 所示,结合子口袋被多个红色高能水合位点占据。例如,#44 水合位点与三唑吡啶酮的羰基碳原子重合,#56 水合位点与三唑吡啶酮片段的甲基重合。在未被化合物 1 占据的子口袋区域,HOH483 与 #4 水合位点重合(ΔG = 1.69 kcal/mol);HOH485 与 #19 水合位点重合(ΔG = 2.26 kcal/mol);HOH486 与 #21 水合位点重合(ΔG = 2.45 kcal/mol)。这一结果表明,在子口袋中引入新的基团以置换 HOH483 和 HOH485 高能水分子,有望提升化合物的结合亲和力。
基于GIST水合自由能解释化合物4与38的活性差异:定性分析
基于水分子热力学分析,Dodda 等人1提出:置换 15-PGDH 子口袋中的高能水分子可带来约 1–2 kcal/mol 的结合自由能增益。将化合物 4(图 2,IC50 = 26 nM)对接至 PDB 条目 9PFL 的结合口袋后发现,其溴原子虽朝向该高能水区域并部分实现水分子置换,但未能形成最优相互作用,因而活性受限。
为充分探索该子口袋的水置换潜力,作者在化合物 4 的骨架基础上,通过 Suzuki 偶联反应设计了 4,846 个虚拟芳基取代衍生物,旨在以不同芳环替代溴原子,从而更有效地置换高能水。随后,结合分子对接虚拟筛选、自由能微扰(FEP)计算优先级排序、可视化检查及综合评估,最终选定 12 个候选分子进行合成。其中,化合物 38(图 2,IC50 = 0.057 nM)的活性较化合物 4 提升逾 400 倍,显著验证了水导向设计策略的有效性。
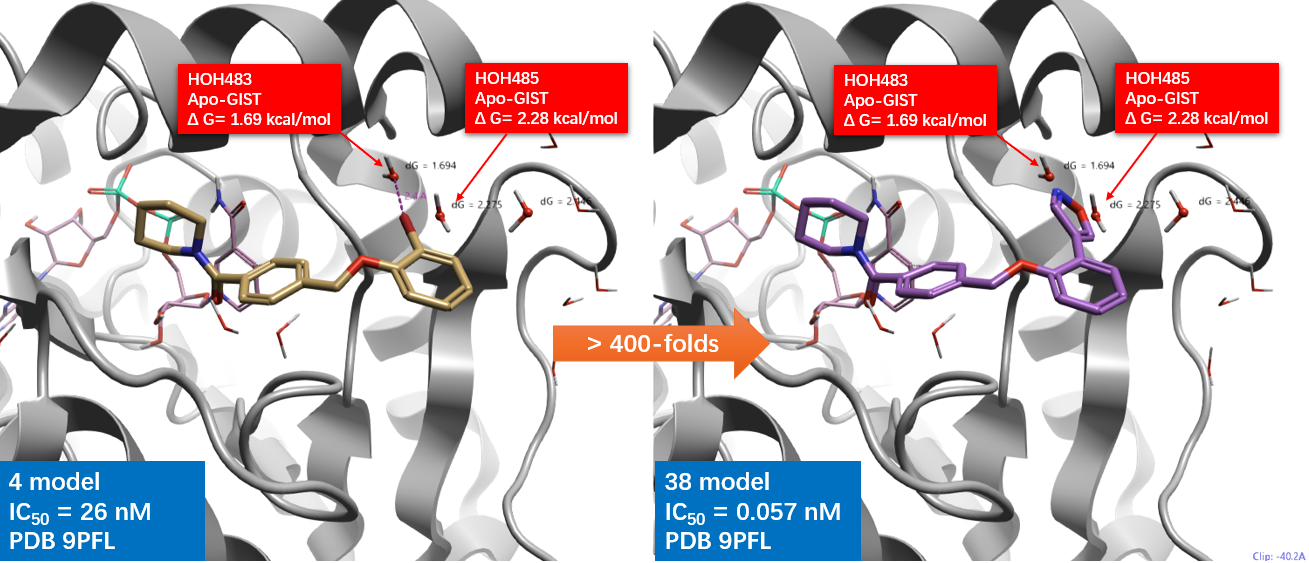
图5. Flare Docking预测的化合物4与38在 15-PGDH(PDB 9PFL)中的结合模式。
采用 Flare Docking(计算引擎为 Lead Finder,设置为“Very Accurate but slow”模式)将化合物 4 与 38 对接至 15-PGDH(PDB: 9PFL)的结合口袋,结果如图 5 所示:化合物 4 的溴原子指向一个高能水合位点,而化合物 38 的异噁唑环则与两个高能水分子位置高度重合。该结果与 Dodda 等人1的对接研究基本一致。

图6. 化合物 4(黄色)与 38(棕色)对接构象叠合比较。网格表面:15-PGDH 蛋白;球体:GIST 预测的高能水合位点;线状结构:共晶结构中的实验水分子。
将两者的对接构象进行叠合(图 6)可进一步揭示其差异:化合物 4 仅有限地延伸至子口袋,对高能水的置换不充分;而化合物 38 则深入占据该区域,完全取代了两个高能水分子。这一观察初步支持了核心假设——化合物 38 因更有效地置换高能水而获得显著的结合自由能优势,从而表现出更高的亲和力。
基于GIST的去溶剂化自由能修正对接打分:定量评估高能水置换的自由能增益
值得注意的是,尽管 Flare Docking 所采用的 LF-dG 打分函数在 CASF2013 基准测试中展现出优异的相关性(Pearson R = 0.66)7,却未能准确区分化合物 4 与 38 的结合自由能。具体而言,Flare Docking 对化合物 4 的预测结合自由能(dG score = –9.93 kcal/mol)与实验值(–10.34 kcal/mol)仅相差 0.41 kcal/mol,预测精度较高;然而,对化合物 38 的预测值(dG score = –9.81 kcal/mol)则显著偏离实验值(–13.96 kcal/mol),误差达 4.15 kcal/mol。
为进一步评估通用打分函数的表现,我们使用 GNINA8(score_only 模式)对上述 Flare 生成的结合构象进行重新打分,结果汇总于表 1。尽管化合物 38 在 gauss_1、gauss_2、疏水项(hydrophobic)及非定向氢键项(non_dir_h_bond)等能量组分上均优于化合物 4,但这些优势未能转化为更高的整体 Affinity 打分。GNINA 预测的结合自由能分别为 –9.53 kcal/mol(化合物 4)与 –10.23 kcal/mol(化合物 38),计算所得 ΔΔG 仅为 0.7 kcal/mol,远低于实验观测值(3.67 kcal/mol)。
| Compound | 4 | 38 |
|---|---|---|
| gauss_1 | 122.46680 | 149.03520 |
| gauss_2 | 1420.47168 | 1643.15222 |
| repulsion | 5.23956 | 6.98573 |
| hydrophobic | 94.15118 | 101.49037 |
| non_dir_h_bond | 1.99749 | 2.97978 |
| Affinity (kcal/mol) | -9.53 | -10.23 |
| CNNaffinity | 6.77 | 7.39 |
| ΔGwatdisp (kcal/mol) | -18.144 | -21.846 |
| Affinity-Corr (kcal/mol) | -9.53 | -13.93 |
| Exp ΔG (kcal/mol) | -10.34 | -13.96 |
与 Flare Docking 类似,GNINA 的 Affinity 打分对化合物 4 的预测较为准确(偏差 –0.81 kcal/mol),但对化合物 38 的预测严重低估其亲和力(偏差达 3.73 kcal/mol)。这一系统性偏差表明,传统对接打分函数难以有效捕捉由高能水置换所贡献的显著自由能增益,凸显了显式水热力学分析(如 GIST)在先导化合物优化中的不可替代价值。
在此,我们引入了基于GIST的配体置换自由能(\(\Delta G_{watdisp}\))来评估配体置换高能水所贡献的自由能增益4,并用来修正分子对接打分:
$$
\begin{aligned}
\Delta G_{\text{bind}}^{\text{docking-GIST-corr}} = \Delta G_{\text{bind}}^{\text{docking}} + \Delta G_{\text{watdisp}} \cdots(1)
\end{aligned}
$$
化合物4与38的\(\Delta G_{watdisp}\)见表1,它们的配体置换自由能差\(\Delta \Delta G_{watdisp}\) 为-3.70 kcal/mol,如公式(2)所示:
$$
\begin{align}
\Delta \Delta G_{\text{watdisp}} &= -21.846\ – (-18.144) \\
&= -3.70 \ \text{kcal/mol} \cdots(2)
\end{align}
$$
修正之后的4与38对接预测值\(\Delta \Delta G\) = 4.4 cal/mol,如公式(3)所示,与实验\(\Delta \Delta G\) = 3.62 kcal/mol 非常接近,仅有0.8 kcal/mol差异。
$$
\begin{align}
\Delta \Delta G_{\text{bind}}^{\text{docking-GIST-corr}} &= \Delta \Delta G_{\text{bind}}^{\text{docking}} + \Delta \Delta G_{\text{watdisp}} \\
&= -0.7 + (-3.70) \\
&= -4.40 \ \text{kcal/mol} \cdots(3)
\end{align}
$$
如表1所示,经过\(\Delta G_{watdisp}\)修正化合物38的预测结合自由能(Affinity-Corr = -13.93),与实验值 -13.96 kcal/mol几乎一致。总的来说,基于GIST的配体置换自由能\(\Delta G_{watdisp}\)可以定量评估化合物4与38在置换高能水带来结合自由能增益上的差异。
FEP+ 未能有效捕捉高能水置换所贡献的结合亲和力增益:基于GIST的配体置换自由能更具解释力
基于化合物 4 的骨架结构,Dodda 等人1通过 Suzuki 偶联反应设计了 4,846 个虚拟芳基取代衍生物,旨在系统探索以不同芳环替代溴原子对子口袋高能水分子的置换能力。首先对全部虚拟化合物进行约束对接筛选,仅保留对接打分优于化合物 4 的约 700 个候选分子进入下一轮评估。随后,从该集合中依据化学多样性原则选取约 70 个代表性化合物,进行自由能微扰(FEP+)计算。利用 FEP+ 预测结果训练随机森林(Random Forest)与 k-近邻(k-NN)回归模型,并用这些模型预测全部 700 个化合物的相对活性。最终筛选出预测活性等于或优于化合物 4 的约 60 个分子,对其对接构象进行人工视觉验证,确保其结合模式在结构上合理且可解释。综合结构合理性、合成可行性及预测活性后,选定 12 个化合物(图 7 中编号 27–38)进行实际合成。

图7. 12 个旨在置换高能水分子的 Region 2 衍生物设计及其对 15-PGDH 的抑制活性(IC50 值)。
图 8 展示了上述 12 个化合物的 FEP+ 预测 pKi 值与实验值的对比关系。根据原文1,其中化合物 31 被定义为“Worst”对照组,化合物 4 为“Parent”,化合物 38 为“Best”。分析表明,化合物 31 并未采取与化合物 4 相似的结合模式,亦未延伸进入子口袋以置换高能水分子,因此本质上属于超出既定 FEP 扰动假设(consistent binding mode)的体系,被作为阴性对照合成。
从统计角度看,图 8 中表面上较高的相关性主要由化合物 31 这一单一数据点所驱动。剔除该不具可比结合模式的数据点后,对剩余 11 个化合物的 FEP+ 预测值(根据图8的坐标轴估计而来)进行统计分析:平均预测值 \(\mu = 8.78\)( pKi),标准差 \(\sigma = 0.44\)( pKi),Pearson 相关性系数 \(R^2 = 0.34\)。该相关性未达到通常认为具有统计意义的阈值(\(R^2 > 0.5\)),且预测值的波动幅度完全落入典型实验误差范围内9。
换言之,在剔除化合物 31 所引入的系统性偏差后,FEP+ 在该体系中的预测结果在统计上难以区分于一个以均值为中心、在实验误差范围内随机波动的“空白模型(null model)”。因此,该 FEP+ 模型在本案例中并未提供超出实验不确定性的有效区分能力,其对先导化合物优化的指导意义有限。
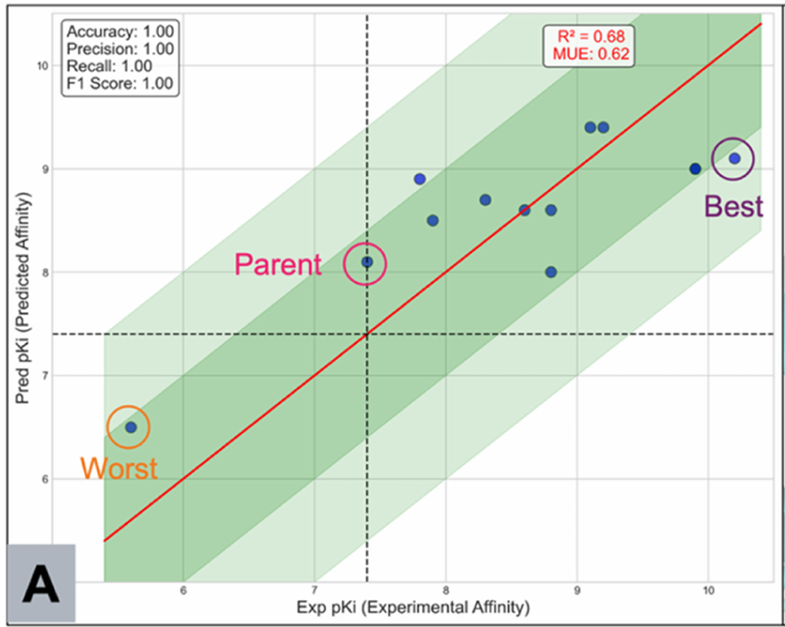
图8.FEP+ 对 12 个衍生物的预测性能评估(数据源自文献 1 图 8)。横轴:实验 pKi;纵轴:FEP+ 预测 pKi。
尽管 FEP+ 在数值上区分了化合物 4(Parent,预测 pKi ≈ 8.11)与化合物 38(Best,预测 pKi ≈ 9.12),对应预测 \(\Delta \Delta G_{\text{pred}} \approx 1.37\ \text{kcal/mol}\),但这一差异远低于实验观测值 \(\Delta \Delta G_{\text{exp}} = 3.62\ \text{kcal/mol}\)。这表明,在本研究体系中,FEP+ 对由高能水置换所驱动的结合自由能增益评估能力有限,其预测精度显著低于基于 GIST 分析所得的水置换自由能贡献项 \(\Delta G_{\text{watdisp}}\)。该结果进一步凸显了显式溶剂热力学建模在识别关键水介导相互作用中的独特价值。
方法
蛋白结构准备
使用 Cresset Flare v10 对 PDB 9PFL 的 A 链进行结构准备(Protein Prep 模块,Normal 模式),包括添加氢原子、优化残基质子化状态以及修复缺失侧链。B 链被移除,其余共晶水分子与其它成分均被保留,以维持结合口袋的原始化学环境。
apo-GIST 计算
为表征配体结合前蛋白结合口袋的溶剂化热力学性质,而非配体诱导的水网络重排效应,本研究采用无配体条件下的 GIST 分析(apo-GIST)。计算仅包含蛋白 A 链及其共晶水分子,并使用原共晶配体定义 GIST 分析区域,以确保关注区域与真实结合口袋一致。
具体 GIST 计算参数如下:
- Calculation method: Normal
- Ligand: None
- Grid spacing: 0.5 Å
- Grid definition: Ligand
- Chains: A Chain, A Water
- GCNCMC: During equilibration only (buffer 4.0 Å)
- Simulation length: 20 ns
- Solvent model: Explicit TIP4Pew water
GIST 结果以 Flare 项目文件形式保存(附件:15-PGDH-apo-gist.flr)。
水合位点的识别与自由能计算
水合位点(hydration sites)基于 GIST 计算得到的水氧密度网格进行识别。具体而言,水合位点采用贪心峰值拾取(greedy peak picking)方法进行识别:首先选取具有最高水氧密度的体素作为第一个水合位点中心,并排除其 2.5 Å 半径范围内的体素;随后对剩余体素重复该过程,直至不存在密度高于体相水氧密度 2 倍的体素。该过程用于识别空间上离散且具有统计意义的高占据水合区域。
对于每一个水合位点,收集其 1.4 Å 半径内的体素,并对其中的 GIST 热力学量进行体积积分。若体素位于水合位点的范德华半径范围内,其对应的能量被累加,并乘以体素体积(0.125 Å3),得到以 kcal/mol 表示的水合自由能。通过pyflare脚本实现,具体方法参见文献 4。
水密度及对应的 ΔG 网格数据以 dx 格式导出,并使用 pyflare 脚本生成水合位点模型,结果以 PDB 格式保存(附件:apo_hs.pdb),其B-factor列为水合自由能。
配体水置换自由能 \(ΔG_{watdisp}\) 的计算
当配体原子占据原本由水分子占据的结合位点区域时,将引起局部去溶剂化效应,从而产生与水置换相关的自由能变化,记为 \(ΔG_{watdisp}\)。该量通过累积配体原子范德华半径范围内体素的 GIST 热力学量,并进行体积积分计算,方法与水合位点自由能计算保持一致(体素体积为 0.125 Å3),具体实现参见文献 4。
需要强调的是,\(ΔG_{watdisp}\) 仅刻画由水分子置换所带来的溶剂热力学贡献,并不包含配体构象应变、蛋白构象重排或直接相互作用能项,因此不能被视为完整结合自由能的替代,而应作为独立的物理修正项使用。
使用 pyflare 脚本分别计算化合物 4 与化合物 38 的 \(ΔG_{watdisp}\),结果见附件。
结论
本研究通过回溯性分析 15-PGDH 抑制剂从化合物 4 到化合物 38 的优化历程,系统论证了结合位点高能水分子在驱动活性跃升中的关键贡献作用,并全面评估了多种计算方法在捕捉此类效应上的能力差异。GIST 分析成功识别出子口袋中多个热力学不利的水合位点(\(\Delta G = 1.69\text{–}2.45\ \text{kcal/mol}\)),为理性设计提供了明确指导。结构叠合与结合模式分析表明,化合物 38 通过异噁唑环完全置换两个高能水,而化合物 4 仅实现部分置换,这一定性差异解释了其 400 余倍的活性提升。
然而,常规计算工具在此场景下表现不佳:主流对接打分函数(比如GNINA)因忽略显式水热力学贡献,严重低估化合物 38 的亲和力;即便采用高精度的 FEP+ 方法,其预测 \(\Delta \Delta G\)(\(\approx 1.37\ \text{kcal/mol}\))仍远低于实验值(\(3.62\ \text{kcal/mol}\)),且整体预测相关性弱(\(R^2 = 0.34\)),表明水置换主导且结合模式发生变化的情形下,FEP+ 的适用性受到明显限制。
与此形成鲜明对比的是,基于 GIST 的配体水置换自由能(\(\Delta G_{\text{watdisp}}\))不仅定性揭示了优化机制,在该体系与优化路径下,更通过简单的线性修正(\(\Delta G_{\text{bind}}^{\text{corr}} = \Delta G_{bind}^{\text{docking}} + \Delta G_{\text{watdisp}}\))实现了对实验 \(\Delta \Delta G\) 的合理定量再现(预测 \(-4.40\ \text{kcal/mol}\) vs. 实验 \(-3.62\ \text{kcal/mol}\))。该结果表明,显式引入水置换相关的溶剂热力学项,能够在该案例中有效补偿传统打分函数对水驱动效应的系统性低估。
综上所述,GIST 不仅是一种有效的水合位点探测工具,更应被视为一种显式引入溶剂热力学信息的物理补充层,弥补现有打分函数与 FEP 方法在高能水置换场景下的不足,而非对其进行替代。本研究表明,将基于 GIST 的水置换自由能项作为独立修正项整合至传统 SBDD 流程中,有助于在保持既有计算框架与效率优势的同时,增强对水介导结合自由能变化的物理可解释性与预测可靠性。
本案例为“以水为中心”的药物设计提供了一条可操作且可扩展的技术路径:在先导化合物优化过程中,应优先识别并针对性占据结合位点中的高能水区域,再结合常规对接或自由能方法对配体–蛋白直接相互作用进行精细化优化,而非仅依赖后者作为唯一设计依据。需要强调的是,上述结论建立在结合模式保持一致、且水置换为主要自由能驱动力的前提之上,其外推性仍有赖于更多体系与设计场景的系统验证。
接下来可以作什么?
- 用Spark水分子替换工作流设计新化合物:在Spark中对高能的水分子进行替换,生成新的化合物。
- 在基于结构的项目中使用预测的水合位点分析、发现新的先导化合物优化契机。
- 在对接中使用GIST的水合位点:ledock与watvina原生支持GIST水和自由能打分,可使用GIST预测的水合位点(b-factor因子为ΔG值)进行分子对接或打分。
- 在对接中使用GIST网格:AutoDock-GIST或DOCK 6可使用GIST水的能量网格与密度网格,增强分子对接性能。
附件
本文计算涉及的关键数据:
1 2 3 4 5 6 7 8 9 10 | . ├── 38.sdf #Flare docking结果 ├── 4.sdf #Flare docking结果 ├── 9pfl_dry.pdb ├── 9pfl_ligand.sdf ├── 9pfl_prot.pdb ├── apo_hs.pdb #apo-GIST水合位点,B-factor为dG值 ├── dG.dx #Flare apo-GIST计算的自由能网格 ├── water_density.dx #Flare apo-GIST计算的水密度 └── 15-PGDH-apo-gist.flr #Flare Project文件,包含上面所有的数据 |
如果需要,请联系我们。
联系我们,获取软件试用
想要尝试Flare信息丰富、用户友好界面,发现它如何帮助您自信地推动潜在先导化合物优化?请现在就联系我们安排试用,快速访问Flare的广泛功能。我们的专业团队随时准备通过安装和设置为您提供支持,而我们全面的教程库——涵盖从常见工作流程到高级方法和功能的所有内容将帮助您开始使用。我们在这里帮助您更快地实现目标,让您设计出感兴趣的分子。
电邮:info@molcalx.com
电话:020 – 38261356
文献
- Dodda, L. S., Campos, S., Ciccone, D., Carreiro, S., Leit, S., Brennan, D., Zephyr, J., Jacques-O’Hagan, S., Kumar, S., Kuo, F.-S., Shaik, M. M., Price, D. J., Loh, C., Edmondson, S. D., Tummino, P., & Kaila, N. (2025). Knowledge and Structure-Based Drug Design of 15-PGDH Inhibitors. Journal of Medicinal Chemistry, 68(17), 18436–18462. https://doi.org/10.1021/acs.jmedchem.5c01231
- Nguyen, C. N., Kurtzman Young, T., & Gilson, M. K. (2012). Grid inhomogeneous solvation theory: Hydration structure and thermodynamics of the miniature receptor cucurbit[7]uril. Journal of Chemical Physics, 137(4), 973–980. https://doi.org/10.1063/1.4733951
- Wahl, J., & Smieško, M. (2018). Thermodynamic Insight into the Effects of Water Displacement and Rearrangement upon Ligand Modifications using Molecular Dynamics Simulations. ChemMedChem, 13(13), 1325–1335. https://doi.org/10.1002/cmdc.201800093
- 肖高铿. 基于GIST的水合位点分析及其在基于结构设计中的应用. 墨灵格的博客. http://blog.molcalx.com.cn/2024/10/04/gist-based-hydration-site-analysis.html
- Flare. https://cresset-group.com/software/flare
- Flare GIST. https://cresset-group.com/software/flare-3drism
- Lead Finder 分子对接性能测试. http://blog.molcalx.com.cn/2018/05/24/leadfinder-evaluation.html
- McNutt, A. T., Li, Y., Meli, R., Aggarwal, R., & Koes, D. R. (2025). GNINA 1.3: the next increment in molecular docking with deep learning. Journal of Cheminformatics, 17(1), 28. https://doi.org/10.1186/s13321-025-00973-x
- Brown, S. P., Muchmore, S. W., & Hajduk, P. J. (2009). Healthy skepticism: assessing realistic model performance. Drug Discovery Today, 14(7–8), 420–427. https://doi.org/10.1016/j.drudis.2009.01.012


