
摘要:本文详细介绍了Flare GIST水分析原理,以及如何进行基于GIST的水合位点分析。GIST分析相比HSA方法的优势在于水密度是水合位点的自然呈现,而无需预测水合位点。HSA方法的优势是直接呈现水合位点的自由能等热力学量,弥补了GIST等值图是分散到每个体素上的贡献值带来不足。本文演示了如何从GIST的水密度开始进行水合位点预测与HSA分析,并将GIST分析与HSA两者联合使用,互补长短、获取更多直观、有价值的信息。本文还列举了几个典型案例,演示了如何在基于结构设计的项目中使用GIST指导分子设计,比如评估结合位点的水热力学特点、预测水合位点、发现关键水分子与相互作用特征以挖掘先导化合物优化机会,作为溶剂置换自由能打分函数对化合物进行排序等等。
肖高铿/2024-10-04
前言
蛋白和小分子的结合过程发生在水溶液中。在体内环境中,蛋白质的活性位点充满了水分子,它们与溶剂中的本体水(bulk water)有着完全不同的热力学性质。当小分子与蛋白结合时,它会导致水分子从活性位点置换到本体区域,这种置换过程的热力学是配体结合自由能的主要来源1-3。
蛋白质周围能量不利的水分子区域被称为“热点(Hot spot)”。我们假设,配体的疏水性部分取代这些不利位置上的水分子将产生对结合自由能有利的贡献,这在疏水性蛋白质环境中的水合位点中是成立的,因为这些位置的水分子不能形成紧密的氢键。相反,对于与蛋白发生强相互作用的水合位点,由于其位于更疏水性的蛋白环境中而具有显著更高的熵,可以用极性的配体片段进行替换。如果配体的极性部分能够以相似的几何形状与蛋白形成相同的相互作用,并且没有熵损失,则它可以提高结合自由能。水分子释放到本体溶液中可能会对亲和力产生净正效应。相反,有利水合位点的特点在于具有潜在的水分子与极性环境形成紧密氢键。应该避免替换这些位置上的水,而如果在几何上可行的话,可以考虑通过保持溶剂分子在位点中的方式来桥接与蛋白的相互作用。无论如何,用极性配体进行替换或与之相互作用时,都必须考虑有利于氢键形成的优势几构型何。
水合位点分析将结合状态的几何构型、水网络与结合自由能联系起来,这有助于揭示结构-活性关系(SAR)的趋势。此外,它还确定了水合位点的置换是焓驱动还是熵驱动地贡献于结合自由能。因此,水合位点与水分子的作用在分子识别的研究中已经得到了广泛的认可4-9。
需要注意的是,对配体结构的每个修饰都会相应地影响各个水合位点以及对能量项的计算。通常人们认为溶剂化能具有可加性,这就像对于定义明确的结合口袋中的官能团,其蛋白质-配体相互作用是独立的一样,然后加和以评估蛋白-配体相互作用能。然而,溶剂效应并不总是局部的10,它有可能是属于一个水网络整体的一部分。水分子在结合位点被配体的一部分置换时,可能会以非加和性的方式改变被置换和未被置换的水分子能量。通常将能量项归属到各个水合位点的水分子可以支持解释,但如果忽视对整个水网络的影响,则可能产生误导。因此,对于所有表征蛋白质中水合位点的方法,重要的是对无配体(apo-)和有配体(holo-)结合的蛋白结构进行分析,以研究更换配体和替换特定官能团对最终水合位点分类的影响。
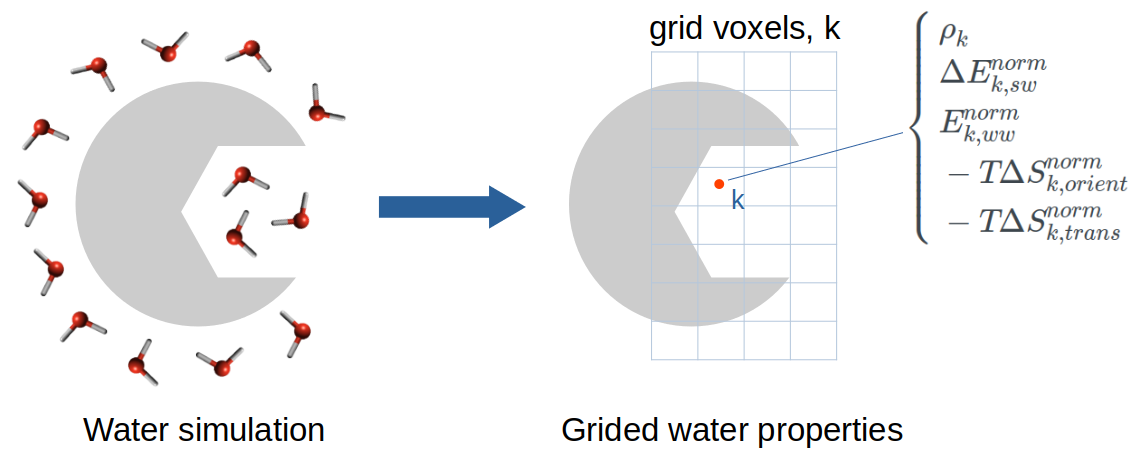
在Flare™中实现的水分析方法之一是由Nguyen等人11提出的GIST(Grid Inhomogeneous Solvent Theory)。它使用显式水对蛋白质进行约束的分子动力学模拟,并使用非均匀溶剂化理论 (inhomogeneous solvation theory,IST) 计算预定义体积(通常是结合位点)的水分子分布和热力学性质。GIST的主要创新之处在于它将IST中的熵和能量表达式出现的空间积分离散化到一个细致、连续的三维网格上。这种离散化处理使得研究者无需预先定义结合口袋中的水合位点或水分子团簇,也不需要对水分子稀少的区域使用另外的理论进行分析。也就是说,GIST将水的性质表示为其空间位置的连续且平滑的函数,而不是IST那样仅仅聚焦于溶剂高密度存在的水合位点。因此在GIST中,水合位点由水分子密度高的区域自然显现。
Flare V9是Flare™系列12软件的最新版本,它为GIST计算结果提供了直观的可视化分析方法,呈现结合位点内水的密度及其热力学性质(自由能、焓与熵),这些信息为基于结构的设计提供了直接的指导。在本站搜索“GIST”可以发现很多用GIST指导设计的案例。尽管Flare提供了便利的GIST分析方法、比IST的水合位点分析呈现了更多的信息,但是还有部分用户希望用显式水分子3D坐标表示的水合位点,并且能够对用其自由能对水分子着色,最后呈现在3D视窗中。因此,本文的主要目的之一是介绍如何用pyflare根据GIST计算结果生成显式的水合位点,并保存为PDB格式,并在其β-factor列填入计算的自由能,并呈现在Flare的3D视窗中进行可视化分析。本文的再一个目的是比较GIST分析与水合位点分析的优缺点,并指出两者是互补地,联合使用会获得更多有用的信息。
方法
Grid Inhomogeneous Solvation Theory (GIST)
网格非均匀溶剂化理论(Grid inhomogeneous solvation theory,GIST)是由Nguyen等人11提出一种强大且易于处理的计算方法,用于计算大分子周围水的水合结构和热力学性质。水分子的热力学性质可以基于非均匀溶剂化理论(IST)使用从显式水和蛋白质的MD模拟获得的轨迹快照计算得到2。除GIST外,大多数其他计算方法都使用水合位点分析(hydration site analysis,HSA)来识别高密度与定域的水,称为水合位点。虽然基于HSA的方法为特定区域水的作用提供了有价值的见解,但它们仍然有一个明显的局限性,即它们不能提供较大的高密度水区域和其他相对于本体而言水密度较低区域的信息13。为了克服这些缺陷,GIST将IST离散化到填充蛋白质活性位点的三维网格上,覆盖所有水的占据区域(图1)。因此,GIST提供了信息更丰富的水合水图像,作为密度分布及其热力学性质的展示。
图1. GIST计算方法示意图:在GIST网格上水的性质是通过蛋白与显式水的分子动力学模拟轨迹计算而来。
GIST计算在感兴趣区域中三维矩形网格中正方体体素(voxel)𝑘上水分子的各种热力学量。GIST方法的完整描述参见原始论文11。在本研究中,我们计算在体素𝑘中水分子的五个GIST性质:
- ρ𝑘, 在体素𝑘中发现的水分子氧原子的密度,单位为本体区(bulk region)密度(本体水的密度ρbulk=1)。
- \(Δ𝐸_{𝑘,sw}^{norm}\), 以本体水(bulk water)为参比,在体素𝑘中每个水分子的溶质-水相互作用的平均能量(kcal/mol/water),在本体区溶质-水相互作用的能量微乎其微,贡献为零。
- \(𝐸_{𝑘,ww}^{norm}\), 在体素𝑘中每个水分子与其他所有水分子的水-水相互作用平均能量的一半(kcal/mol/water)。1/2因子防止了两次水-水相互作用被重复计算,并保留了净水的总能量,即单个水能量的总和。
- \(−𝑇Δ𝑆_{𝑘,orient}^{norm}\), 以本体水(bulk water)为参比(即本体水的取向熵设置为0),在体素𝑘中每个水分子的一阶取向熵(kcal/mole/water)。
- \(−𝑇Δ𝑆_{𝑘,trans}^{norm}\), 以本体水为参比(即本体水的平动熵设置为0),在体素𝑘中每个水分子的一阶平动熵(kcal/mole/water)。
基于这些计算的量,水分子的热力学性质由以下方程描述。本文将相互作用能视为焓的贡献,在体素𝑘中水分子相对于本体(bulk)水的总焓定义为:
$$
Δ𝐻_{𝑘}^{norm}=Δ𝐸_{𝑘,sw}^{norm𝑘}+2\times(𝐸_{𝑘,ww}^{norm}−𝐸_{bulk,ww}^{norm}) \cdots(1)
$$
其中,\(𝐸_{bulk,ww}^{norm}\)表示在本体(bulk region)中水-水相互作用的平均能量。\(Δ𝐻_{𝑘}^{norm}\)表示水分子与蛋白质和所有其他水分子相对于本体水\(2𝐸_{bulk,ww}^{norm}\)的平均相互作用。相似地,在体素𝑘中的水分子相对于本体水的总熵定义为:
$$
−𝑇Δ𝑆_{𝑘}^{norm}=−𝑇Δ𝑆_{𝑘,orient}^{norm}−𝑇Δ𝑆_{𝑘,trans}^{norm} \cdots(2)
$$
其中𝑇是绝对温度(默认情况下包含在GIST的熵项中)。因此,在体素𝑘中水分子相对于本体(bulk)水的自由能是总焓与熵的和,写为:
$$
Δ𝐺_{𝑘}^{norm}=Δ𝐻_{𝑘}^{norm}−𝑇Δ𝑆_{𝑘}^{norm} \cdots(3)
$$
然后,不利的水分子(unfavorable/unhappy water)具有正的自由能(\(ΔG_{𝑘}^{norm}>0\));相反,有利的水分子(favorable/happy water)具有负的自由能(\(ΔG_{𝑘}^{norm}<0\))。如上所述,这些热力学量代表了与本体水的热力学量的差值,这意味着对高自由能水的置换是蛋白质配体结合的驱动力。
GIST在蛋白质-配体结合和配体设计中的应用仍在探索中。在本文中,我们用Flare V9的GIST水分析12功能来获得上述的热力学量,比如水的密度、自由能、焓与熵的贡献。
Flare GIST水分析
以PDB 3SV2与2ZF0的“湿”结合位点apo结构(不包含配体与金属离子)的GIST分析为例,在蛋白下载到Flare V912,然后进行结构准备,最后共晶结构的A链水包含在GIST分析里面,具体的GIST分析条件如下:
- Calculation method: Normal
- Ligand: None
- Grid spacing: 0.5 Å
- Grid Definition:Ligand
- Chains: A Chain, A Water
- Simulation length: 20ns
- Solvent Model: explicit TIP4Pew Water
在这个计算中,“湿”结合位点意味着在分子动力学模拟之前将共晶结构的水链包含在GIST分析里。计算完毕,将水密度(Water density)以及ΔG(方程3)、ΔH(方程1)、-TΔS(方程2)等热力学量导出为dx格式文件用于一下步的水合位点识别与分析。
如果没有特别说明,其它蛋白的GIST分析也用相同的参数,包括对包含配体的holo-GIST分析也是如此。
水合位点的识别及其热力学性质计算
水合位点依赖于高密度水分子的位置,使用水的氧密度网格进行识别。在本文中,采用常用的贪心峰值拾取(greedy peak picking)法对密度网格进行聚类:首先,选择具有最高水氧密度的体素来确定第一个水合位点的位置。然后排除所有在第一个水合位点2.5Å范围内的体素,不再考虑。然后,对下一个最高密度的体素重复此过程,直到没有剩下密度高于2倍本体水氧密度的体素为止。
水合位点的自由能根据其所占据的体素累积而来。首先识别每个水合位点周围1.4Å半径内的体素(Voxel):
$$
V = \left\{v_{i} | v_{i} \in hydration\ site\right\} \cdots(4)
$$
如方程4所示,如果一个体素包含在水合位点的范德华半径内,则累加这些体素的能量,并将总和乘以体素的体积(vol = 0.125 Å3),得到以kcal/mol为单位的值,如方程5所示:
$$
ΔG = vol \times \sum_{v_{i} \in V}G_{GIST}(v_{i})\cdots(5)
$$
GIST是一种基于网格的方法,它使用分子动力学(MD)模拟来计算蛋白质表面上每个水分子的位移成本。在这个过程中,可能会遇到一些具有极高或极低能量的体素,这些体素可能代表了非常紧密或非常松散地与蛋白质表面相互作用的水分子。这些极端值似乎反映了用于GIST计算的约束分子动力学模拟的特性,因为当在分子动力学模拟中允许侧链移动时,这些极端值会大大减弱或完全消失。因此有必要将那些具有极高或极低能量值的体素在计算自由能时被忽略或被限制在一个合理的范围内,以避免它们对整体计算结果产生过大的影响。
在不同的文献报道中,会使用不同的截断值策略来消除极端值的影响。比如,在Balius等人14研究中用3 kcal・mol-1・Å-3限截断值来限制极高能量值的体素,这个值比平均体素能量值高出12个标准差。然而使用截断值并不总是对虚拟筛选的性能有好处,在测试的25个体系中,有的体系改善、有的体系变差。在Uehara等人18的研究中,设置下限截断值为1 kcal・mol-1・Å-3来去掉贡献不显著的体素。最近Eberhardt等人19还报道了Gaussian加权缓冲处理的方法,使用σ值进行高斯加权计算(其中σ = r/3,r为半径)来计算每个水合位点的热力学量,高斯加权确保了靠近水合位点中心的体素比远离的体素有更大的权重。最后,将GIST网格能量值乘以体素体积(0.125 ų)转换为kcal/mol。
在计算的时候,对体素vi上的GGIST值设置了下限截断值±0.5kcal・mol-1・Å-3来去掉贡献不显著的体素,使用上限截断值+3kcal・mol-1・Å-3去掉极端高值的体素,如方程6所示:
$$
G_{GIST}(v_{i}) = \left \{
\begin{aligned}
& 0\ &if\ |G_{GIST}(v_{i})| \lt 0.5 \\
& +3\ &if\ |G_{GIST}(v_{i})| \ge +3 \\
& G_{GIST}(v_{i}) &\ otherwise
\end{aligned}
\right.\cdots(6)
$$
在本文中,我们用pyflare脚本实现四种计算结果:1)仅使用下限截断值;2)仅使用上限截断值;3)同时使用了下限截断值与上限截断值的结果;4)不使用任何截断值。为了与不使用截断的结果相比,将使用截断值的结果标注为_trunced。
方程(5)计算的ΔG为将水合位点的水从结合位点移到本体水所需克服的能量,并未考虑移除之后其周围环境可能发生的重组。
方程(5)地通过pyflare脚本实现,利用Flare GIST得到的水密度(Water density)与水自由能(ΔG)格点文件来生成水合位点,同时计算水合位点的自由能。
配体结合的置换自由能:ΔGwatdisp
配体结合到蛋白的过程中,配体置换出结合口袋里的水。为了定量了解配体原子占据水分子位置的去溶剂化自由能,需要求解被占据格点上自由能的累加值。
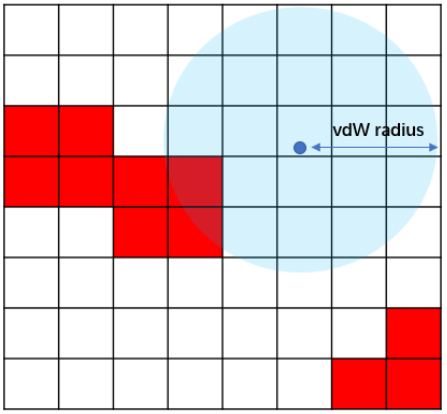
图2. 用GIST来计算当配体原子占据结合水分子的位置时发生的蛋白去溶剂化自由能。蓝色球:配体的原子;网格:体素。
当配体原子占据了结合位点里水分子的位置时,会发生对应的蛋白去溶剂化自由能变化,配体获得对应的结合自由能增益,表示为ΔGwatdisp。为了评估受体在结合过程中的去溶剂化自由能,首先识别被配体原子置换的体素(Voxel),如方程7所示:
$$
V = \left\{v_{i} | v_{i} \in ligand\right\} \cdots(7)
$$
图2解释了计算方法,如果一个体素包含在一个配体原子的范德华半径内,则认为该体素被配体置换。累加这些体素的能量值,并将总和乘以体素的体积,得到以kcal/mol为单位的值,如方程8所示:
$$
ΔG_{watdisp} = \alpha \times vol \times \sum_{v_{i} \in V}G_{GIST}(v_{i})\cdots(8)
$$
其中α是缩放因子。在Balius等人14的研究中,探索了多种α值,当α=-0.5时,比起没有使用的基于GIST蛋白去溶剂化的打分方法,大部分情况下虚拟筛选的性能得到显著地提高。而在本文中,如果没有说明则α=-1。其中,在GIST计算时格点为0.5 Å(见方法部分,space=0.5 Å),因此这里的vol = 0.125 Å3。此外,在计算的时候,对体素vi上的GGIST值设置了方程(6)的上限与下限截断值。一个体素可能被一个配体的多个原子置换,但在计算自由能贡献时,一个体素仅贡献一次。
需要注意的是,方程(8)是(5)的反向符号版本(同时计算时的原子半径也不同),这意味着并未考虑置换之后周围环境的变化以及对水网络的破环等因素影响。
方程(8)的计算方法通过pyflare脚本实现,可利用Flare GIST得到的水自由能(ΔG)格点文件来计算结合配体的水置换自由能。
结果
BTK抑制剂8的水分子替换实验
Smith等人成功地将基于片段的药物设计应用于非共价键结合的BTK抑制剂发现15。化合物2(见表1)是最令人感兴趣的化合物,因此对之进行了结构优化。在2的8位引入不同片段以便探讨构效关系。初步的SAR探索直接发现了化合物8(表1), 比2显著地提高了活性与选择性。
表1. 片段苗头化合物2的SAR探索15
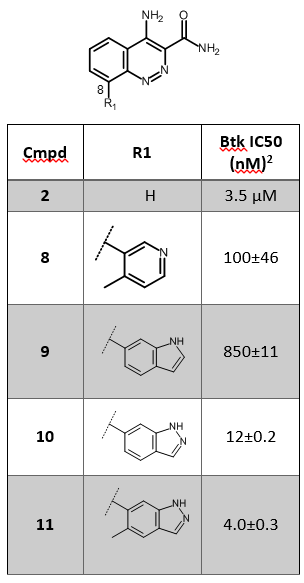
化合物8与BTK的X-衍射晶体结构数据(PDB 4ZLZ)表明:在活性位点里有一个水分子介导了吡啶环N与P-Loop骨架上Phe413以及GLY414残基之间的氢键相互作用15(图3)。将3位取代的4-甲基吡啶片段用小的双环杂环替换这该水分子、并使得新化合物直接与P-Loop区直接发生氢键作用,结果发现了化合物10与11(表1)。比之起始化合物,用这个水分子替换策略设计的新化合物10与11对BTK的活性约提高了10倍。
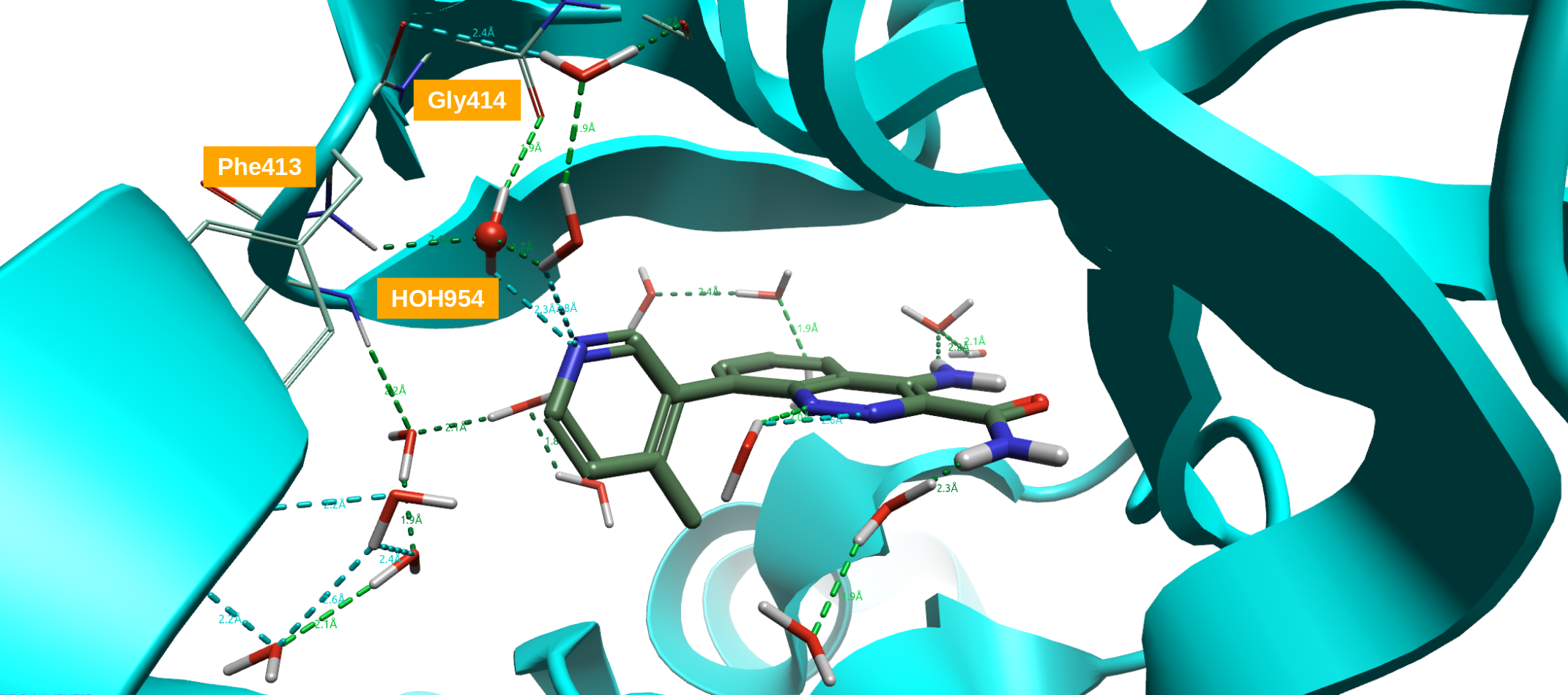
图3. 化合物8与BTK的共晶结构(PDB 4ZLZ):水分子954介导了吡啶环N与P-Loop骨架上Phe413以及GLY414残基之间的氢键相互作用
apo-GIST分析
对化合物8与BTK的共晶复合物结构(PDB 4ZLZ)的结合位点进行apo-GIST分析,观察如图4所示的Water density\(\ge\)4等值图,可以发现水密度图与配体关键的重原子、以及结合位点里靠近配体的结合水位置基本重合,其中包括我们关心的HOH954。这体现了GIST计算方法比IST方法的创新之处:水密度是水合位点的自然呈现。
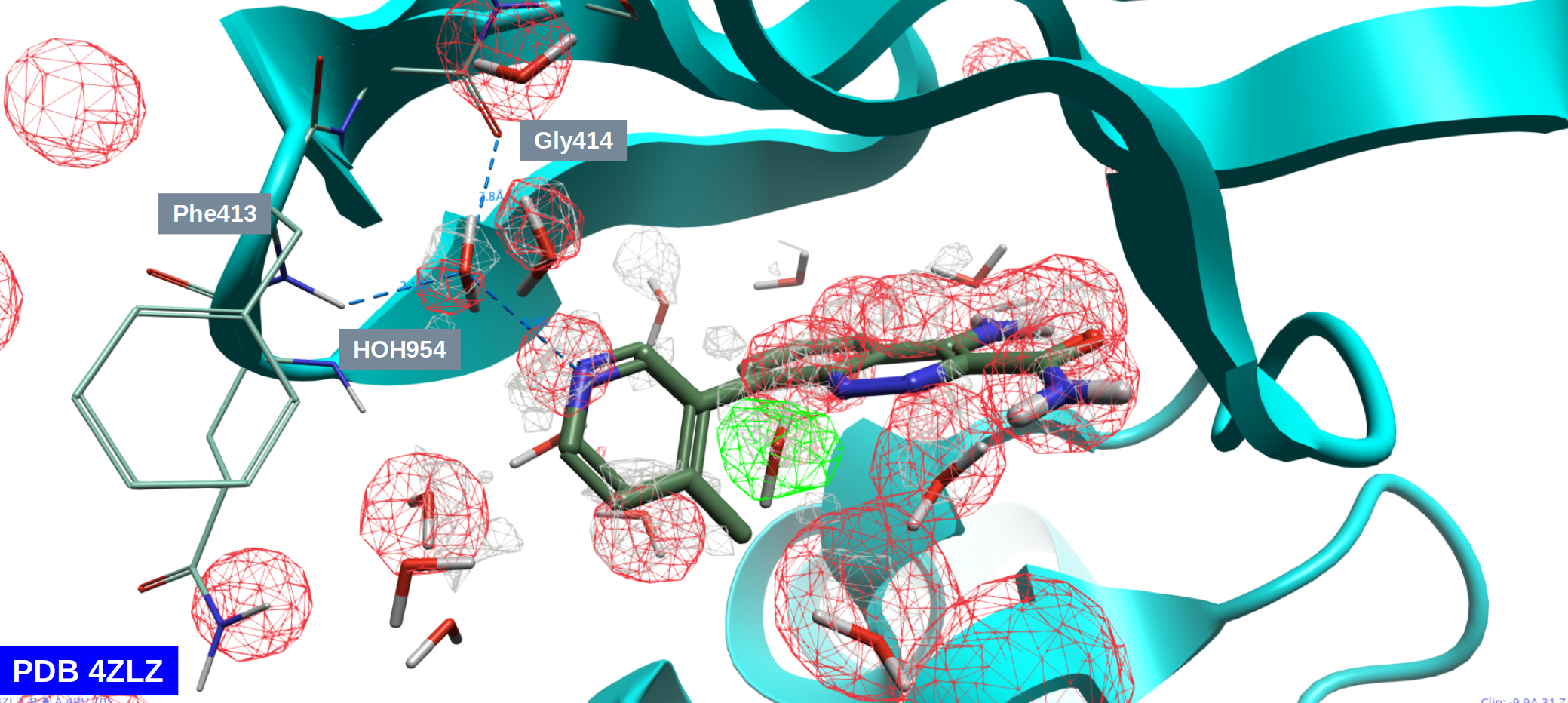
图4. 对PDB 4ZLZ结合口袋的apo-GIST分析结果。灰色网格:density\(\ge\)4的水密度图;红色网格:ΔG\(\ge\)2.0kcal・mol-1;绿色网格:ΔG\(\le\)-1.0 kcal・mol-1。
进一步观察红色网格(unhappy)的水合自由能ΔG\(\ge\) 2.0 kcal・mol-1的等值图,可以发现HOH954被红色网格覆盖,是一个介导蛋白-配体氢键网络的高能水,这对HOH954进行水分子替换设计的决策起到至关重要的作用。
注意:在对GIST的结果进行3D可视化分析时,热力学性质ΔG有两种可能:
- 方程(3)的\(ΔG_{𝑘}^{norm}\):在体素k上单位体积的自由能(kcal・mol-1・Å-3)
- 方程(5)的\(ΔG\):即水合自由能(kcal・mol-1)
一般情况下,GIST方法默认绘制的是方程(3)表示的在体素k上单位体积的自由能,而在图4中绘制的是方程(5)计算的水合自由能。
计算结合水的自由能及其可视化分析
在确定了对HOH954感兴趣之后,将距离其1.4Å的体素收集起来,共有94个体素,其中48个体素的值不为0:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 | x y z distance dG dG_trunced ------- ----- ------ -------- ------ ---------- -25.921 6.849 -8.818 1.164 0.011 0.011 -25.921 6.849 -8.318 1.011 0.096 0.096 -25.921 7.349 -8.318 0.998 0.017 0.017 -25.421 6.349 -8.818 1.077 0.013 0.013 -25.421 6.349 -8.318 0.910 0.093 0.093 -25.421 6.349 -7.818 0.998 0.016 0.016 -25.421 6.349 -7.318 1.290 0.016 0.016 -25.421 6.849 -8.818 0.797 1.461 1.461 -25.421 6.849 -8.318 0.550 16.328 16.328 -25.421 6.849 -7.818 0.686 1.570 1.570 -25.421 6.849 -7.318 1.067 0.008 0.008 -25.421 7.349 -8.818 0.781 0.292 0.292 -25.421 7.349 -8.318 0.526 4.222 4.222 -25.421 7.349 -7.818 0.667 0.427 0.427 -25.421 7.349 -7.318 1.055 0.017 0.017 -24.921 6.349 -8.318 0.781 0.020 0.020 -24.921 6.349 -7.818 0.881 0.017 0.017 -24.921 6.349 -7.318 1.202 -0.003 -0.003 -24.921 6.849 -8.818 0.645 0.239 0.239 -24.921 6.849 -8.318 0.289 2.035 2.035 -24.921 6.849 -7.818 0.501 0.705 0.705 -24.921 6.849 -7.318 0.959 0.081 0.081 -24.921 7.349 -8.818 0.625 0.228 0.228 -24.921 7.349 -8.318 0.241 1.421 1.421 -24.921 7.349 -7.818 0.475 0.271 0.271 -24.921 7.349 -7.318 0.945 -0.000 -0.000 -24.921 7.849 -9.318 1.303 0.009 0.009 -24.921 7.849 -8.818 0.930 0.095 0.095 -24.921 7.849 -8.318 0.730 0.026 0.026 -24.921 8.349 -8.818 1.356 0.126 0.126 -24.921 8.349 -8.318 1.227 0.002 0.002 -24.921 8.349 -7.818 1.294 0.015 0.015 -24.421 6.349 -7.318 1.314 0.017 0.017 -24.421 6.849 -7.818 0.730 -0.004 -0.004 -24.421 6.849 -7.318 1.095 -0.006 -0.006 -24.421 7.349 -8.818 0.819 0.002 0.002 -24.421 7.349 -8.318 0.582 0.007 0.007 -24.421 7.349 -7.818 0.712 0.017 0.017 -24.421 7.349 -7.318 1.084 0.007 0.007 -24.421 7.849 -8.818 1.070 0.607 0.607 -24.421 7.849 -8.318 0.902 0.091 0.091 -24.421 7.849 -7.818 0.990 0.000 0.000 -24.421 8.349 -8.318 1.337 0.098 0.098 -24.421 8.349 -7.818 1.398 0.016 0.016 -23.921 7.349 -8.818 1.205 -0.000 -0.000 -23.921 7.849 -8.818 1.388 0.081 0.081 -23.921 7.849 -8.318 1.263 0.011 0.011 -23.921 7.849 -7.818 1.327 0.006 0.006 Total unique voxels: 48 dG : 3.853 kcal/mol dH : -1.042 kcal/mol -TdS: 4.895 kcal/mol |
用方程(5)计算的溶剂化自由能、焓、熵的值分别为3.853、-1.042与4.895 kcal/mol。这是一个焓有利、熵不利的高能“unhappy”结合水,这与它作为水桥介导蛋白-配体的氢键相互作于的特点是一致的。用方程(6)定义的下限截断值去掉贡献值小的体素,并用上限截断值约束过高的极端值,结果如下:
1 2 3 4 5 6 7 8 9 10 11 12 13 | x y z distance dG dG_trunced -------- -------- -------- -------- --------- ---------- -25.4211 6.84932 -8.8184 0.79702 1.46056 1.46056 -25.4211 6.84932 -8.3184 0.55031 16.32840 3.00000 -25.4211 6.84932 -7.8184 0.68589 1.56977 1.56977 -25.4211 7.34932 -8.3184 0.52646 4.22188 3.00000 -24.9211 6.84932 -8.3184 0.28939 2.03515 2.03515 -24.9211 6.84932 -7.8184 0.50134 0.70519 0.70519 -24.9211 7.34932 -8.3184 0.24096 1.42149 1.42149 -24.4211 7.84932 -8.8184 1.07037 0.60691 0.60691 Total unique voxels: 8 dG : 3.544 kcal/mol dG_trunced: 1.725 kcal/mol |
结果表明,在HOH954 1.4Å的范围之内,共有8个体素满足方程(6)的下限截断值,根据方程5加和法计算的溶剂化自由能ΔG=3.54 kcal/mol。将体素上的值超过方程(6)上限的体素取截断值,则计算的溶剂化自由能ΔG_trunced=1.73 kcal/mol。因此如果对HOH954进行置换,预期可获得 ~1.73 kcal/mol的结合自由能增益。
方程(5)的计算结果也相当容易可视化。以配体周围结合水为例,将计算自由能保存在PDB的β-factor列,即可对结合水按自由能着色,以便快速发现令人感兴趣的可置换高能水。图5展示了配体周围4Å内的结合水,可以明确地让人注意到介导配体与蛋白氢键相互作用的HOH954,其自由能为3.54kcal/mol。
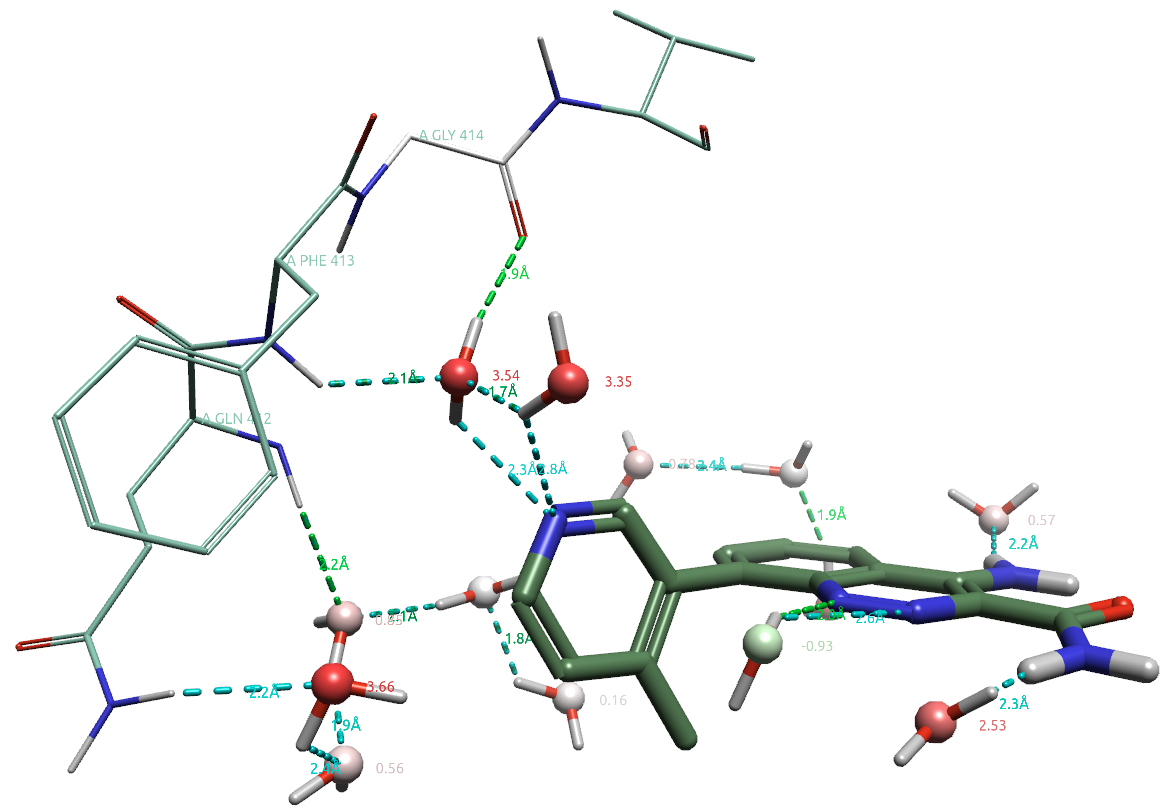
图5.化合物8(PDB 4ZLZ)周围4Å内结合水及其自由能。其中水的氧原子数值为自由能,颜色根据自由能大小着色。
对特定位置结合水的自由能计算与可视化分析是GIST分析的非常重要补充,因为GIST可视化分析仅是呈现分散到每个体素上的值,虽然这样可以突显重要的水,但是没有给出该水的溶剂化自由能(也就是附素值的和)。
水合位点分析(Hydration site analysis,HSA)
水合位点依赖于高密度水分子的位置,可以使用水的氧密度网格进行用聚类算法识别17。首先,选择具有最高水氧密度的体素来确定第一个水合位点的位置。然后排除所有在第一个水合位点2.5Å范围内的体素,不再考虑。然后,对下一个最高密度的体素重复此过程,直到没有剩下密度高于2倍本体水氧密度的体素为止。这里采用2.5 Å进行聚类而不是Young等人17所述的1.0 Å,主要是考虑到水分子的半径1.4Å,这决定了两个水分子间距在2.3~3.3 Å(容差d=0.5Å)时是可以互相融洽地接触。一方面,从合理区间取了个数学上容易计算的2.5(Å),另一方面这个值接近理想值2.8Å。此外,聚类时采用的水密度下限截断值为2,而不是Young等人17的1。还根据方程(5)计算了预测的水合位点的自由能。
图6展示了在PDB 4ZLZ结合位点里距离共晶配体4Å内预测的水合位点(球状)、实验解释的结合水(细棒状)以及water density=2、4的等值图(分别为灰色与蓝色网格图)。可以发现,大部分识别出的水合位点落在water density=4的等值图里,少部分落在water density=2的等值图里。大部分实验结合水都与识别的水合位点一致,配体关键的杂原子也有识别的水合位点与之重合。其中红色虚线圆圈高亮显示的结合水HOH954与识别的水合位点几乎完全一致。
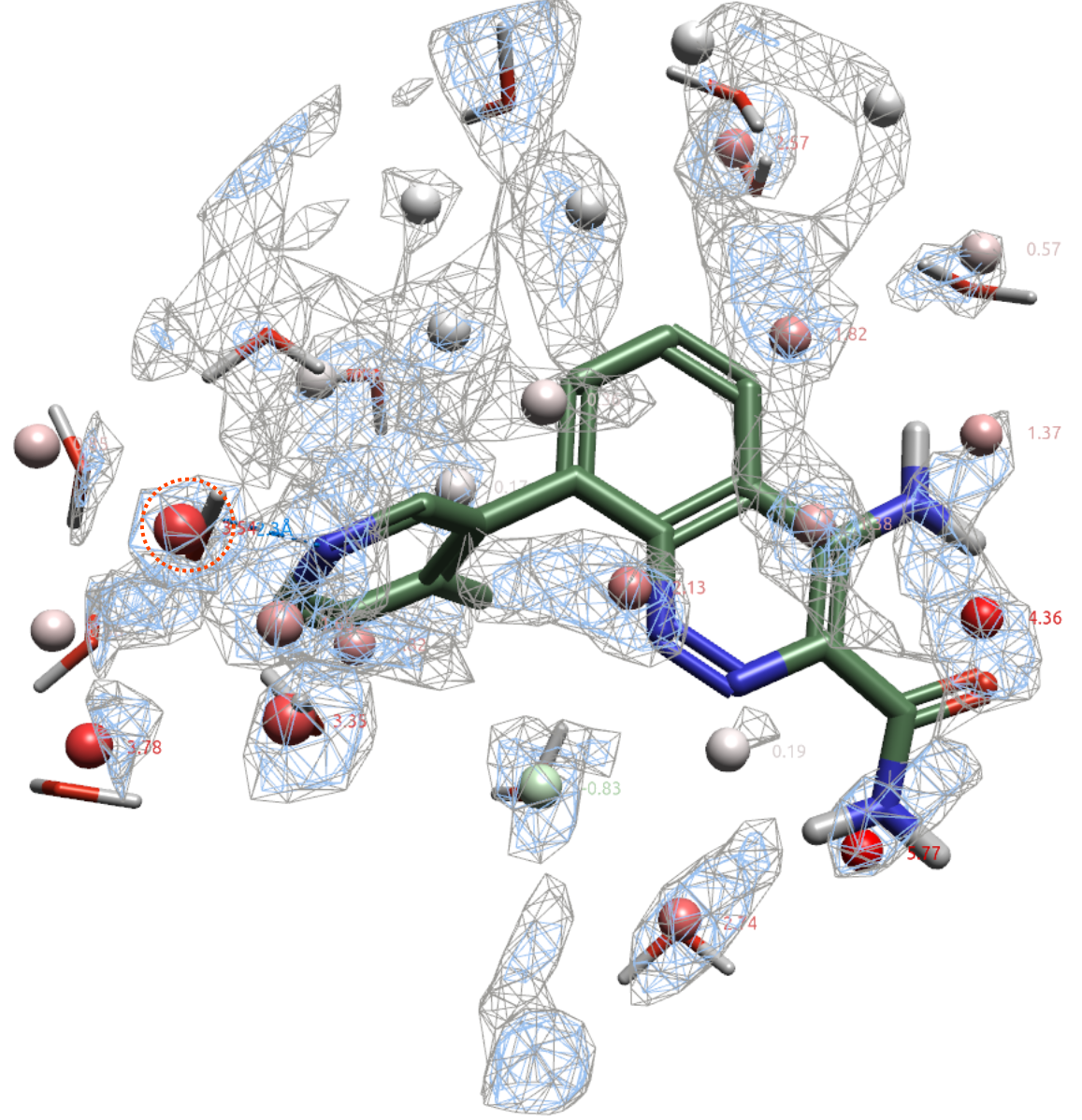
图6.化合物8(PDB 4ZLZ)周围4Å识别的水合位点及其自由能。灰色网格:Water density=2;蓝色网格:Water density=4;球:识别的水合位点,颜色根据自由能大小着色;细棒状:实验结合水。其中计算识别的水合位点上数值为自由能。
比较最高能与最低能的6个计算识别的水合位点HS1-6(图7-左)与晶体结构里结合水W1-6(图7-右)可以进一步凸显预测效果,它们不仅在位置上一致,而且在自由能上也一致。其中桥接蛋白与配体氢键的结合水HOH954(W1)对应预测的HS1,它们的自由能均为3.54kcal/mol。
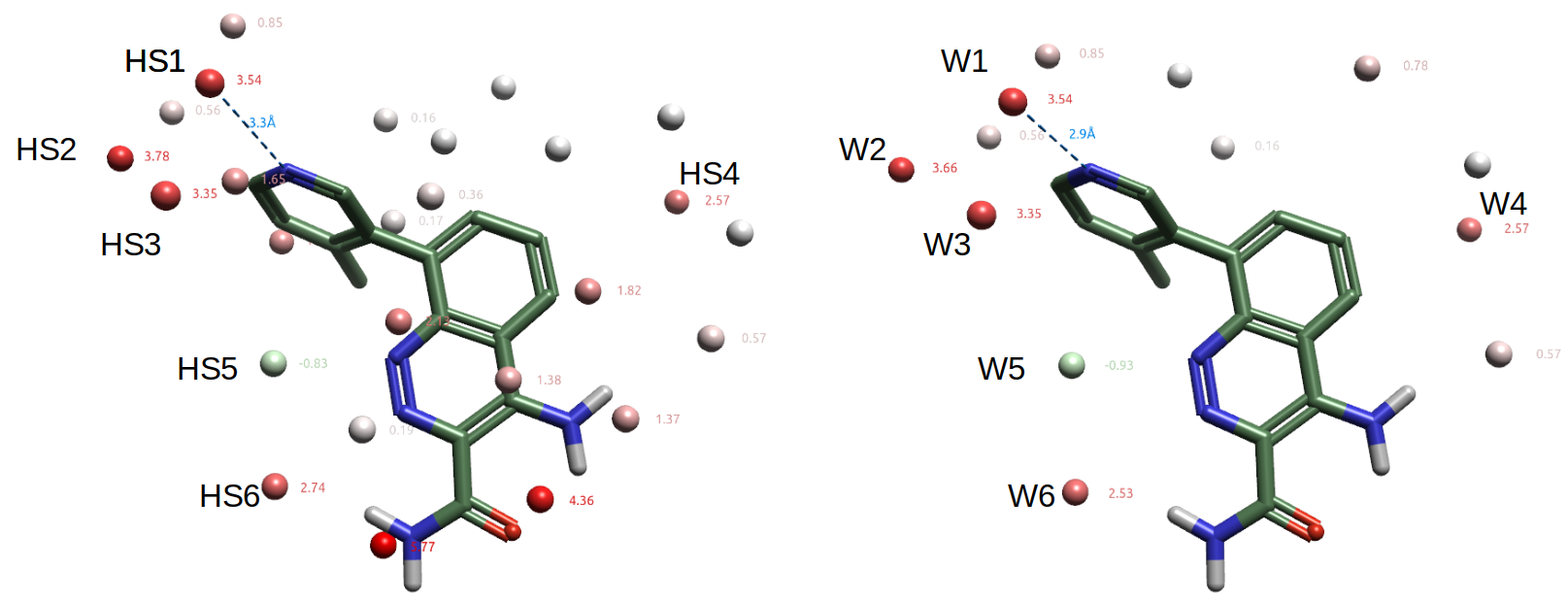
图7. 关键的水合位点(左)与实验结合水(右)的比较。其中氧原子上呈现的数值为ΔG(未使用上限截断)
根据GIST的水密度预测水合位点,并用GIST热力学网格计算水合位点的自由能,这样的水合位点分析(Hydration site analysis,HSA)方法弥补了GIST可视化分析所缺的水合位点自由能,而GIST的可视化分析使得水分子的位置具有动态的特点。HSA与GIST可视化分析是互补的两种方法。
解释SAR
如前所述,想要对抑制剂8进行结构修饰以替换HOH954而获得预期的结合自由能增益,还要求化合物以优势的几何与蛋白形成氢键相互作用,也就是要求新化合物与蛋白在形状与静电上互补,这一点在前文16已经讨论过,这里仅作简要总结。
如图8所示,化合物9虽然与起始化合物(这里把8与HOH954的合并看成一个整理)在形状上相似,但是化合物9没有模仿出HOH954的氧负静电场,因此不能与蛋白发生静电互补、模仿与HOH954-蛋白相似的氢键相互作用,9不仅不能获得预期的结合自由能增益,而且活性还降低了。
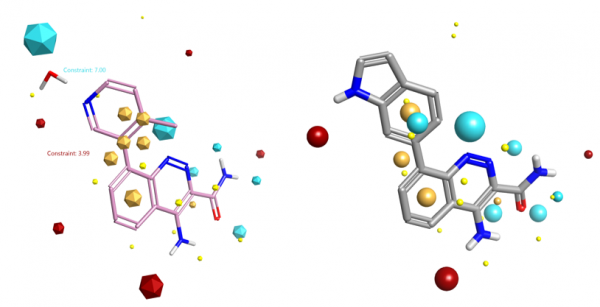
图8.化合物9(右)与起始分子(8与HOH954的合并)的配体场点比较
如图9所示,化合物10、11不仅与起始分子(化合物8与HOH954的合并)在形状上相似,而且在静电上也相似,这确保了10、11能够与蛋白发生静电互补,模仿了HOH954与蛋白发生的氢键相互作用。因此10、11获得预期的结合自由能增益,ΔΔG实验值分别为1.26、1.91 kcal/mol,这与计算的预期值1.73~3.54kcal/mol基本一致。
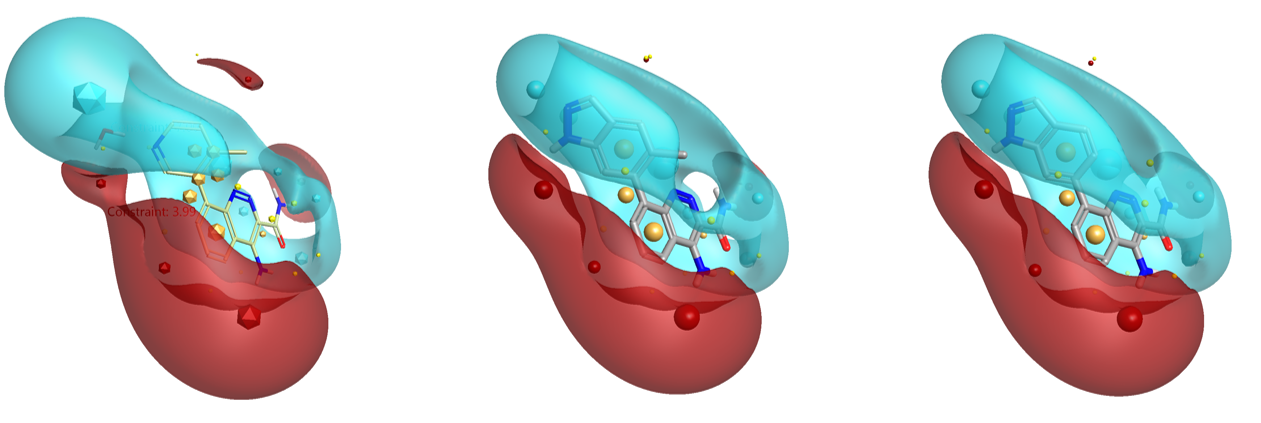
图9. 左边:起始分子的静电(8与HOH954合并);中间:化合物11(Btk IC50 = 4.0 nM)的静电;右边:化合物10 (Btk IC50 = 12 nM)的静电。场/场点颜色编码: blue = negative; red = positive; yellow = steric; gold = hydrophobic。
基于GIST的去溶剂化打分
比之IST方法,GIST方法的另一个优点是可以避开水合位点的预测而直接用热力学性质网格计算配体结合过程中的蛋白去溶剂化自由能项11。已经有很多文献描述此类基于GIST的专项打分函数,这里不在叙述。基于GIST的蛋白去溶剂化打分已经整合到分子对接软件DOCK3.714与AutoDock-GIST18,并在测试中表现虚拟筛选的性能提高。
配体结合过程的蛋白去溶剂化自由能项也称为置换自由能,表示为方程(8)的ΔGwatdisp。在前文20中已经通过几个PDE10A抑制剂的苗头化合物优化充分演示了该方法的应用,这里进行简要的描述。
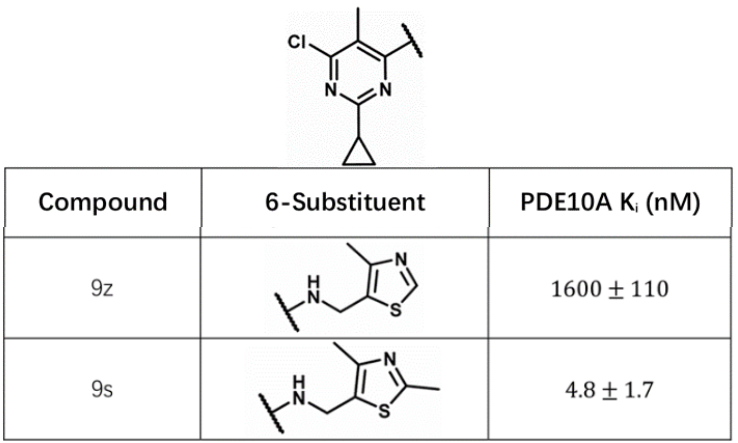
图10. PDE10A抑制剂苗头化合物9s与9z的化学结构及其活性
两个PDE10A抑制剂的苗头化合物9s与9z的化学结构如图10所示,这里存在活性悬崖现象:9s仅比9z多了一个甲基,而结合亲合力活性提高了330多倍(ΔΔG = 3.44 kcal/mol)。初步可以将这个活性差异归结为9s对结合位点深处的结合水HOH1015(见图11)的置换。
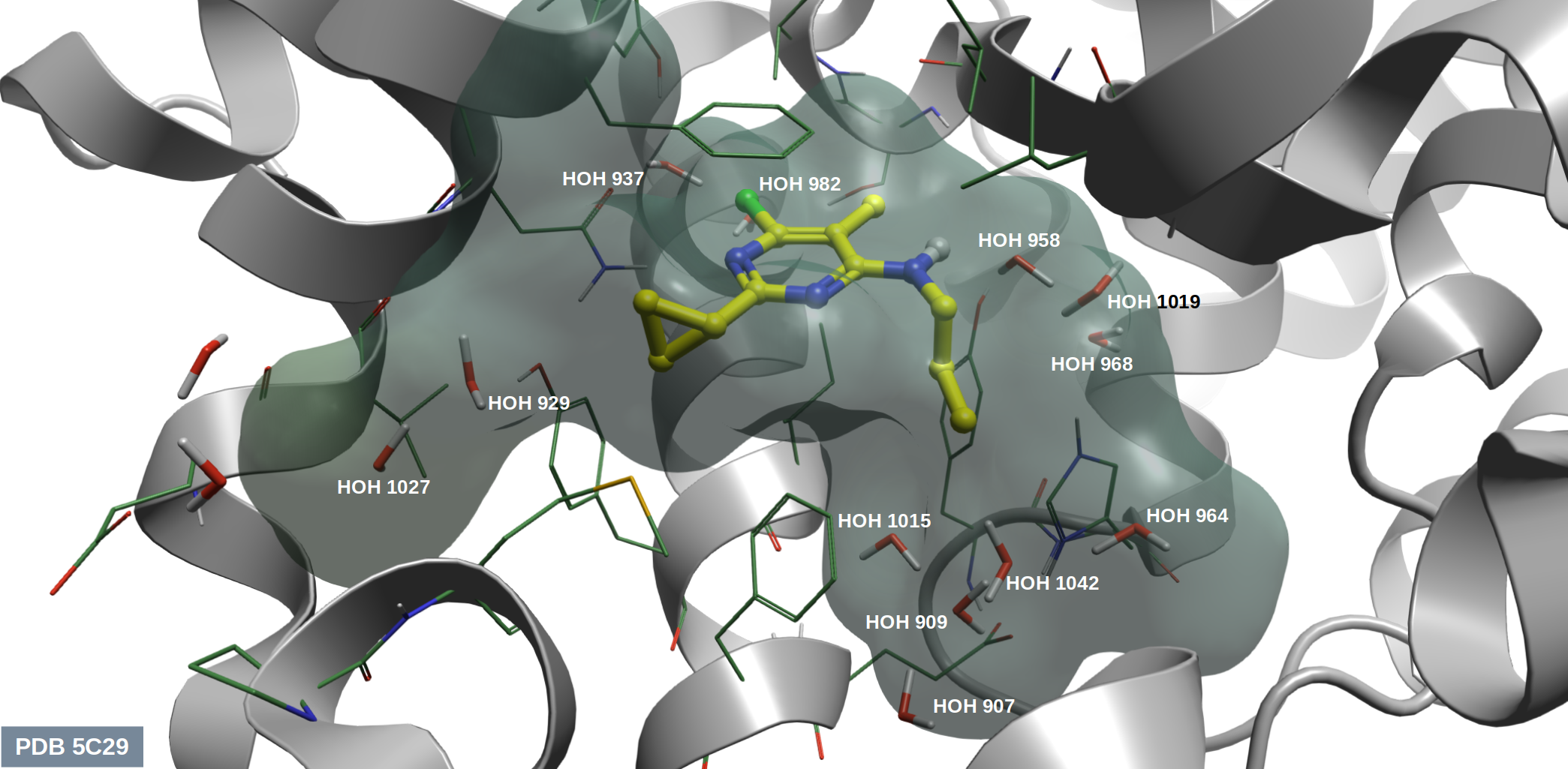
图11. PDE10A的结合位点及其溶剂
用Flare对PDE10A(PDB 5C29)进行apo-GIST计算,然后用方程(5)计算HOH1015的溶剂化自由能,并用方程(8)方法计算9s与9z的ΔGwatdisp,结果如表2所示。
表2. ΔGwatdisp的计算值、及其变化与实验值的比较
| Items | ΔGwatdisp | ΔΔGwatdisp | Exp. ΔΔGbind |
|---|---|---|---|
| HOH1015 | 5.00* | - | - |
| 9s | -41.60 | 3.37 | 3.44 |
| 9z | -38.23 |
单位:kcal/mol;*:溶剂化自由能
结果表明,距离HOH1015水分子1.4 Å范围之内共有40个体素,其中下限截断值高于0.5kcal/mol的体素共18个:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 | x y z distance dG dG_trunced -------- --------- -------- --------- ----------- ------------------- 10.19521 13.17006 41.7274 1.39791 5.19446 3.00000 10.19521 13.17006 42.2274 1.25321 2.33735 2.33735 10.19521 13.67006 41.7274 1.25108 3.21279 3.00000 10.19521 13.67006 42.2274 1.08702 0.83338 0.83338 10.69521 12.67006 42.2274 1.28051 0.79780 0.79780 10.69521 13.17006 41.7274 1.06506 20.36070 3.00000 10.69521 13.17006 42.2274 0.86646 7.79934 3.00000 10.69521 13.67006 41.2274 1.27633 2.22786 2.22786 10.69521 13.67006 41.7274 0.86337 17.14060 3.00000 10.69521 13.67006 42.2274 0.60151 5.86556 3.00000 10.69521 14.17006 41.7274 0.92546 1.19197 1.19197 11.19521 12.67006 41.7274 1.30518 1.86438 1.86438 11.19521 13.17006 41.2274 1.30314 1.33371 1.33371 11.19521 13.17006 41.7274 0.90253 6.82475 3.00000 11.19521 13.17006 42.2274 0.65648 2.68514 2.68514 11.19521 13.67006 41.2274 1.14421 1.21156 1.21156 11.19521 13.67006 41.7274 0.65240 4.87310 3.00000 11.19521 13.67006 42.2274 0.20500 1.47881 1.47881 Total unique voxels: 18 dG : 10.904 kcal/mol dG_trunced: 4.995 kcal/mol |
用方程(5)加和法计算这些体素的自由能贡献为10.9kcal/mol,按方程(6)取上限截断值后为5.00 kcal/mol。无论如何,这提示了置换HOH1015水分子可能获得大幅度的自由能增益。
用GIST ΔG网格对化合物9s,9z打分,发现9s比9z因为引入甲基取代基而置换了HOH1015多获得-3.44kcal/mol(ΔΔGwatdisp)的结合自由能,这与实验结果-3.77 kcal/mol一致!在这个例子中,GIST计算不仅指出了优化方向,而且对化合物进行了精确的排序。
识别关键相互作用,指导新分子设计
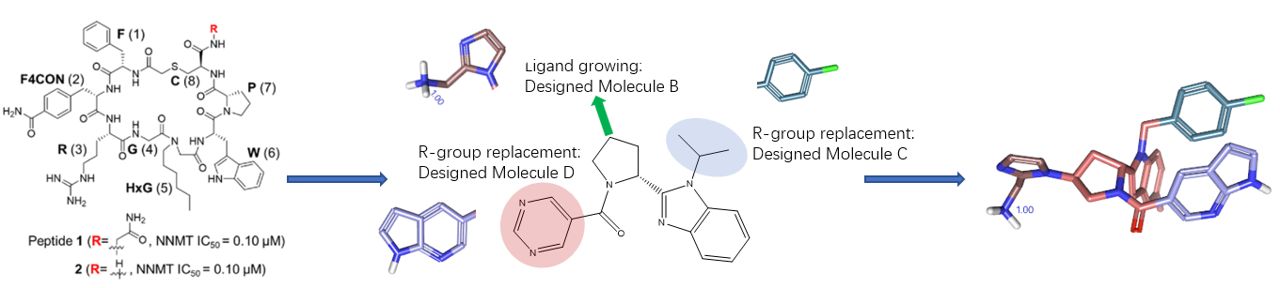
图12. 从肽到小分子的先导化合物优化策略
水分析结果有助于确定抑制剂的潜在药效团,一方面可以用水合位点来约束、优选药效团特征21,另一方面水合位点是潜在的药效团特征22。田野义制药公司的Yoshida等人23提出了一种新的从肽类到小分子先导的发现策略:结合大环肽展示筛选快速识别高亲和力肽类配体,然后用药效团指导从头小分子设计来克服肽类配体的成药性问题。Yoshida等人23就是通过GIST水分析从肽类抑制剂中识别关键的药效团特征,用来指导苗头化合物到先导化合物的优化。我们用Flare GIST与Spark采用同样的策略,如图12所示,从苗头化合物开始,往三个方向生长侧链,重现了Yoshida等人的先导化合物生成策略24。
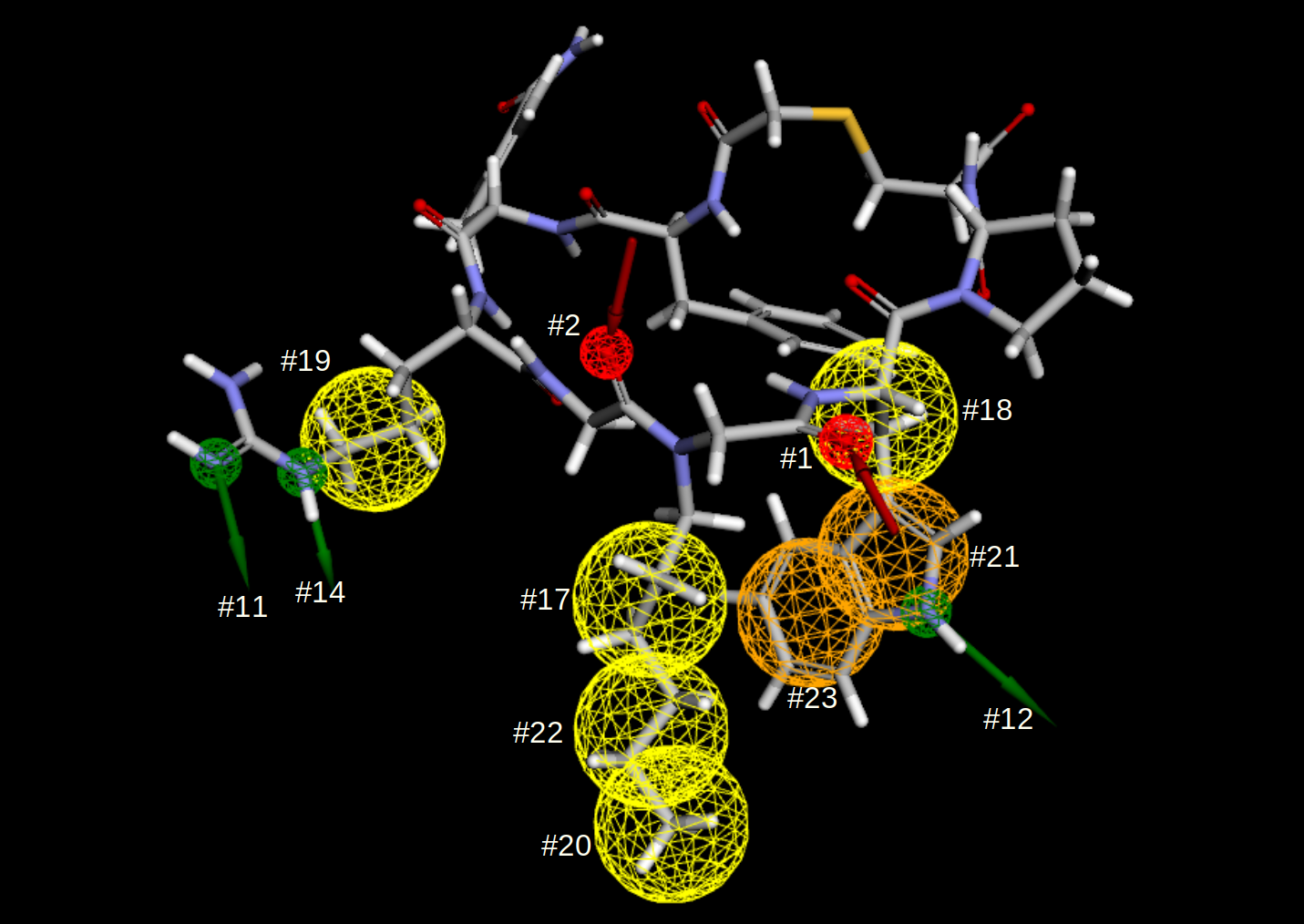
图13. PDB 7WMC揭示大环肽的抑制剂与蛋白相互作用的药效团。绿色箭头:氢键供体;红色箭头:氢键受体;亮黄色的球:疏水相互作用;金黄色球:芳香相互作用。点击图片可动画展示:按住鼠标左键拖动进行转动,按住鼠标右键拖动进行放大与缩小。
其中关键的一点是,我们可以用GIST的网格对环肽与靶标的共晶结构初步揭示的相互作用特征(图13)进行打分,以定量考察药效团特征所在位置的热力学性质。下面是用GIST自由能网格计算主要特征位点的溶剂化自由能:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 | feature_id feature_name x y z dG dG_trunced ---------- ------------ ------- ------ ------ ---------- ------------------ 1 HBA_target -4.492 27.915 38.440 1.582 1.340 2 HBA_target -5.449 25.806 34.032 16.786 5.605 11 HBD_origin -5.498 26.879 25.636 5.901 4.222 12 HBD_origin -6.759 32.246 38.851 2.796 2.656 14 HBD_origin -7.343 27.832 26.601 1.208 1.208 17 H -4.594 30.660 33.953 2.344 2.344 18 H -7.879 28.502 38.508 1.896 1.896 19 H -8.766 27.739 27.713 2.513 2.513 20 H -3.646 34.779 33.784 1.250 1.250 21 AR -7.426 31.217 38.285 1.455 1.455 22 H -4.183 33.019 33.642 3.165 3.165 23 AR -7.947 32.414 36.294 3.365 3.365 |
将大环肽苗头化合物及其场点结合GIST水合位点一起分析,如图14所示,有助于进一步对结合模式进行约束,创建更有意义的药效团模型用于虚拟筛选或用来作为后续Spark的约束条件。
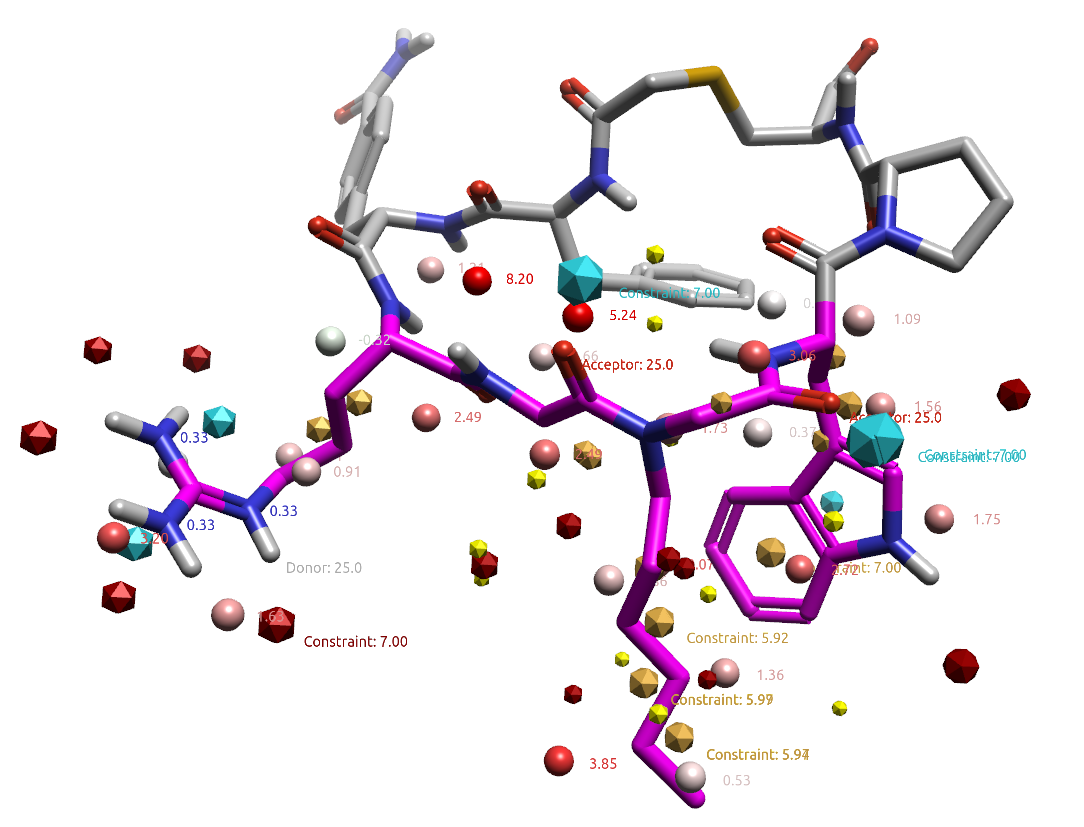
图14. 大环肽及其场点、水合位点。场点用八面体表示,其中金黄色:疏水,红色:正静电势,蓝色:负静电势;圆球:GIST水合位点;棍棒:大环肽。
以大环肽的正己烷侧链为例,在图13中用3个疏水特征#17、20与22来描述,这与图14的侧链疏水场点表示也非常一致,它们所在位点水合自由能分别为2.34、1.25与3.17kcal/mol,此外侧链还与高能的水合位点重合,说明此处引入疏水基团进行水分子替换对活性有利。与正己烷侧链连接的大环肽骨架上,距离最近的两个酰胺羰基氧作为氢键受体(图13药效团特征#1、2),它们所在坐标的水合自由能分别为1.34与5.61 kcal/mol,也分别与两个水合位点重合。苗头化合物Hit A的分子对接结果表明,打分最佳的结合模式其疏水的苯并咪唑与疏水特征#17、20与22(图13)重合,而咪唑N上的异丙基与芳香性疏水中心#23(图13)重合,异丙基的两个甲基分别置换了两个高能水合位点,羰基氧与嘧啶氮原子作为氢键受体与氢键受体特征#1、2(图13)重叠的很好,如图15所示。
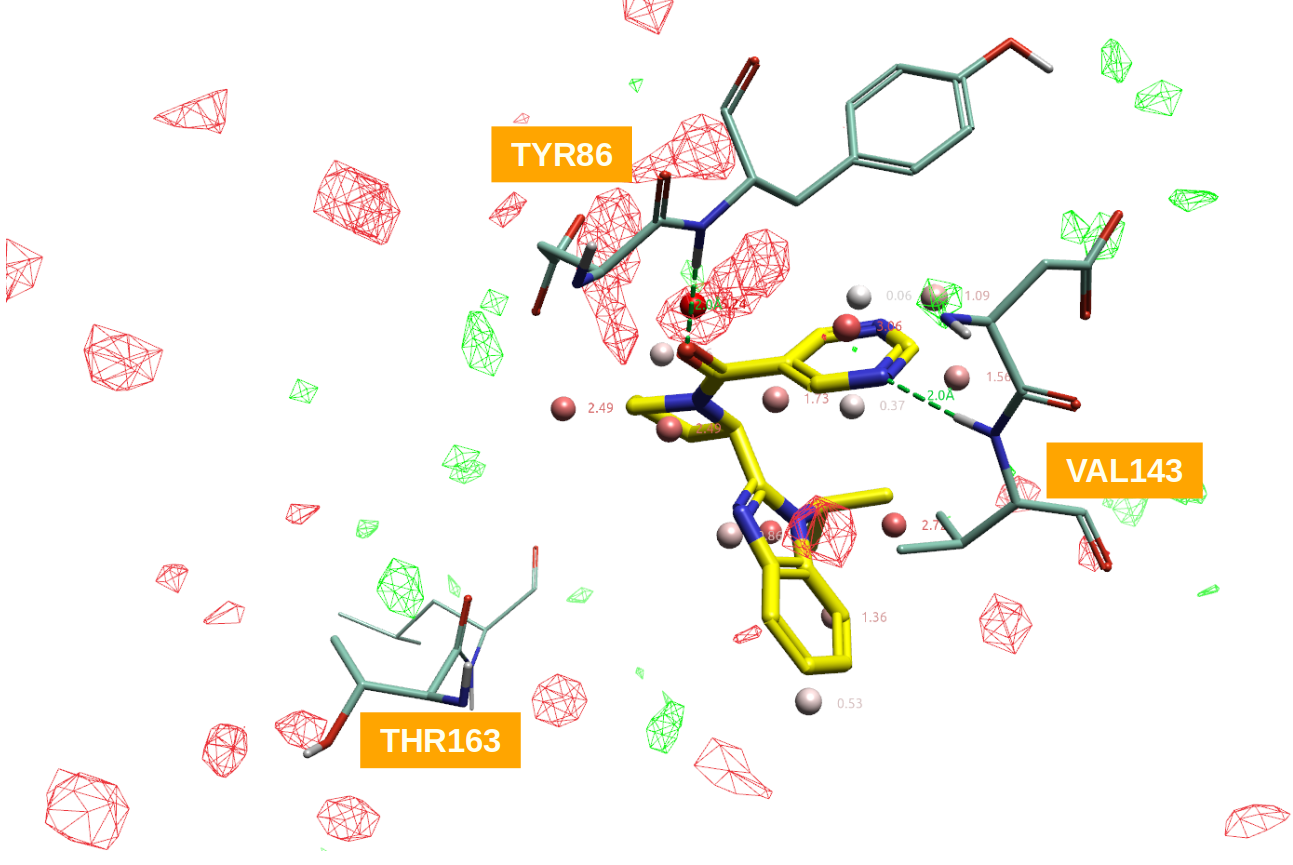
图15.苗头化合物Hit A预测的结合模式,水合位点与GIST ΔH等值图。红色网格:GIST \(ΔH \gt 2.0 kcal・mol^{-1}・Å^{-3}\)等值图;绿色网格:GIST \(ΔH \lt -2.0 kcal・mol^{-1}・Å^{-3}\)等值图;球状:水合位点(数值为水合自由能);棍棒:Hit A。点击图片可动画展示Hit A与药效团叠合结果:按住鼠标左键拖动进行转动,按住鼠标右键拖动进行放大与缩小。
总的来说,根据GIST计算的水合位点可以合理的解释Hit A对接结合模式。一方面,Hit A的羰基氧与嘧啶氮氢键受体与高密度、高能的水合位点重合;另一方面,Hit A的N-异丙基苯并咪唑片段的苯基与异丙基上的两个甲基这两个疏水片段所在位置具有GIST描述的疏水特征:不仅与高能(\(ΔG>0\))的水合位点一致,而且是焓不利\(ΔH>0\)的。这为继续往三个方向进行分子生长设计提供了坚实的基础与信心。更详细的内容,请参阅前文23。
设计卤键相互作用
Wang等人25对卤键数据集研究的表明:参与卤键相互作用的卤原子在配体结合后经常置换/替换计算的结合位点水。这为卤键的引入提供了指导,提高卤键设计的成功率。
David Becherer等人26发现了系列CD38抑制剂,部分化合物的结构与活性如图16所示,它们涉及水分子置换与卤键相互作用,这里用来回溯性地讨论如何用GIST分析结果理性指导卤键设计。
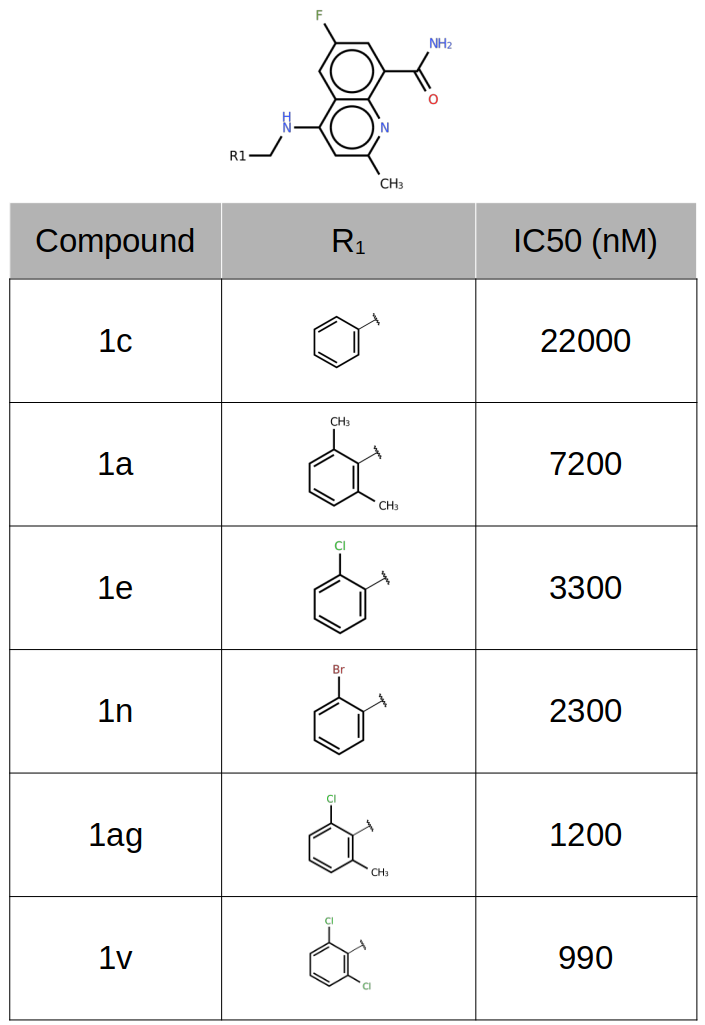
图16. CD38抑制剂的化学结构
用Flare分析CD38与抑制剂共晶结构PDB 4XJS的结合口袋,计算apo-GIST水合位点以及XED vdW相互作用势场(PIP)。结果如图17所示,其中飘带代表靶标CD38,棒状分子为抑制剂1e(图16),红、绿球形分子为GIST水合位点(数值为水合自由能),红、绿网格代表GIST ΔH=+/-1 kcal/mol的等值图(红色为正值,绿色为负值),金黄色等值图为XED力场计算的蛋白vdW相互作用势(PIP)。
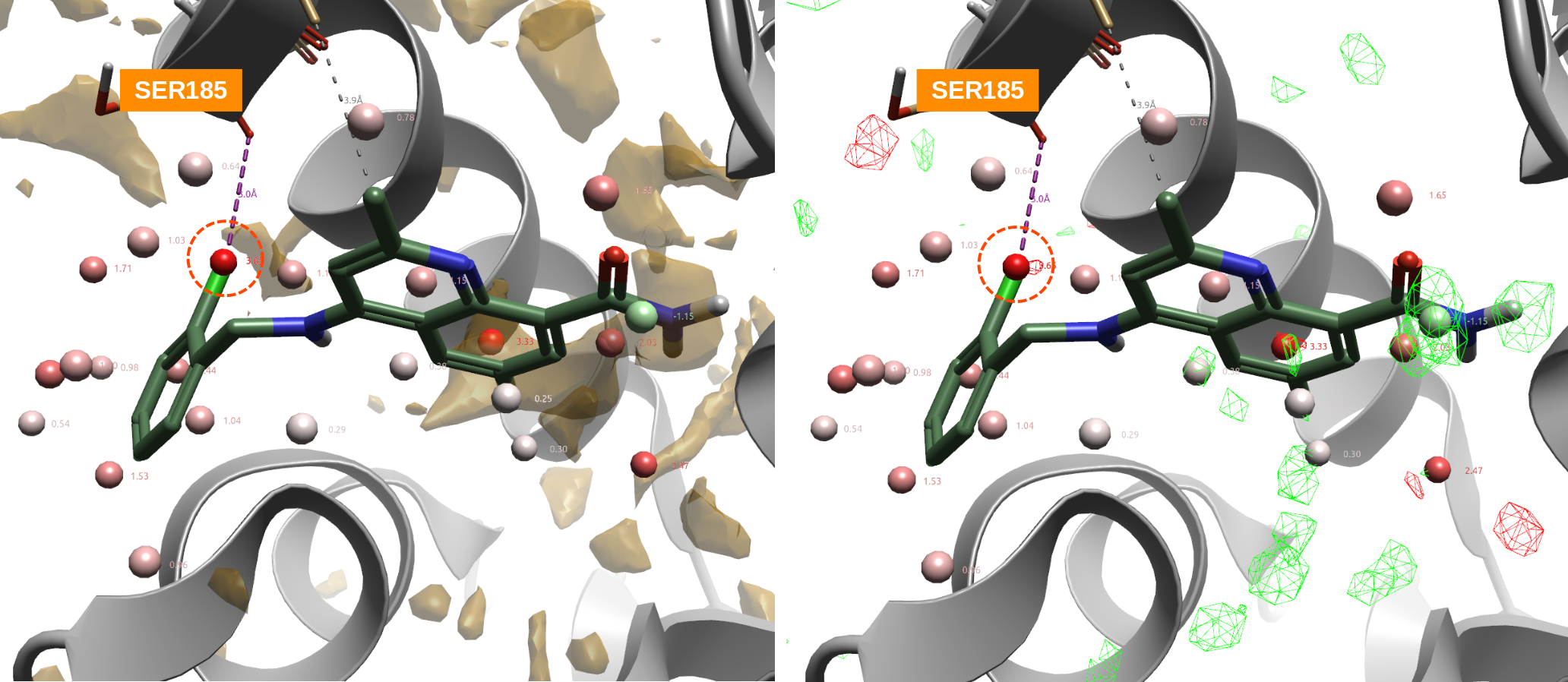
图17. CD38与抑制剂共晶结构PDB 4XJS的apo-GIST分析结果。其中飘带状:CD38;棒状分子:抑制剂1e;红、绿球形分子:GIST水合位点(数值为水合自由能;红、绿网格:GIST ΔH=+/-1 kcal/mol等值图,红色为正值,绿色为负值;金黄色等值图:蛋白XED vdW相互作用势(PIP)。
17图-左显示了XED力场计算的apo-“干”蛋白结合位点vdW PIP(金黄色等值图)与GIST计算的水合位点。分析红色圆圈高亮显示的那个水合位点GIST热力学量,结果如下:
1 2 3 4 | dG : 3.686 kcal/mol dH : 1.824 kcal/mol -TdS :1.862 kcal/mol dG_trunced: 3.655 kcal/mol |
该位点不仅是高能(ΔG=3.65 kcal/mol)、焓不利(\(ΔH>0\)),并与vdW PIP = 3 kcal/mol等值图重合,说明该位点是疏水基团有利、同时也是vdW相互作用有利的位点。
17图-右显示了水合位点以及GIST ΔH等值图(红色为焓不利区;绿色为焓有利区)。17图-右红色圆圈高亮显示的水合位点水的GIST ΔH为红色,因此为焓不利区,这与该水合位点ΔH=1.82kcal/mol是一致的。结合蛋白vdW PIP、GIST ΔG、ΔH信息,说明在该水合位点处引入vdW和/或疏水基团是对配体结合有利的。图17的化合物1e,用氯原子置换高能水合位点,不仅是vdW有利、疏水,而且还有蛋白SER185的羰基氧发生有利的卤键相互作用。
图18. CD38与抑制剂共晶结构PDB 4XJS的apo-GIST分析结果。其中飘带状:CD38;棒状分子:图16的抑制剂;红、绿球形分子:GIST水合位点(数值为水合自由能;红、绿网格:GIST ΔH=+/-1 kcal/mol等值图,红色为正值,绿色为负值。
图18比较了CD38抑制剂R1苯环上不同取代的系列化合物,用vdW PIP与GIST水分析可以解释它们的SAR。
化合物1a(IC50=7200nM)是活性最低化合物1c(IC50=22000nM)的邻位甲基取代衍生物。1a的一个甲基置换了高能(ΔG=3.65 kcal/mol)的水合位点水(图17高亮显示),并且被红色(正值)不利的GIST ΔH等值图覆盖,说明该水合位点用疏水性基团置换是有利的,这解释了1a比1c活性的增加。
而化合物1e、1n、1ag、1v不仅用疏水性卤素置换了高能的水合位点水,而且卤素原子比1a的甲基具有更有利的vdW相互作用,此外还与SER186的羰基氧发生了有利的卤键相互作用,这导致化合物活性进一步提高。这与Wang等人26对卤键数据集研究的结论一致:参与卤键相互作用的卤原子在配体结合后经常置换(发生率=91%)/替换(发生率=82%)计算的结合位点水。
而邻位双取代的1ag与1v比单取代的1e、1n活性更强则与更稳定的结合构象、更高的卤键形成概率有关,请参见博客关于strain enerngy的讨论,这里不再叙述。
总的来说,蛋白PIP分析与GIST水分析为引入卤键相互作用以提高活性的设计提供了有力的工具,也将复杂难以理解的SAR简化。
结论
本文详细介绍了Flare GIST水分析原理,以及如何进行基于GIST的水合位点分析。本文展示了GIST分析相对于IST HSA方法的优势:水密度是水合位点的自然呈现,而无需预测水合位点。本文也展示了HSA方法的优势:直接呈现水合位点的自由能等热力学量,弥补了GIST等值图是分散到每个体素上的贡献值而带来的不足。将GIST分析与HSA两者联合使用可以获取更多直观、有价值的信息。最后,展示了GIST的计算结果直接作为溶剂置换自由能打分函数,在基于结构设计的项目中用来指导分子设计,对化合物进行排序。
代码
水合位点生成:gist_hydrate_sites
依赖Flare python环境的命令行工具,使用方法如下:
1 2 3 4 5 6 7 8 9 10 11 12 13 | (notebookenv) gkxiao@master$ gist_hydrate_sites --help
usage: gist_hydrate_sites [-h] -d <density> -dg <free energy> [-min_dens <min density>] -oprefix <output prefix>
GIST-based Hydration site prediction.
options:
-h, --help show this help message and exit
-d <density> water density in dx format
-dg <free energy> water free energy file in dx format
-min_dens <min density>
min water density,default is 2.0
-oprefix <output prefix>
output prefix |
请联系我们,获取脚本。
用GIST分析感兴趣位置的自由能
方程(5)计算自由能的Flare python GUI代码参见:https://github.com/gkxiao/waters
用法:
- 点击 Flare | Python | Python Interpretor |Load ..导入脚本,见图19步骤1, 2与3。
- 点击感兴趣的蛋白,确保已经计算过GIST,见图19步骤4。
- 点击一个感兴趣的原子,点击Run按钮,计算该原子所在位置的GIST溶剂化自由能,见图19步骤5、6。
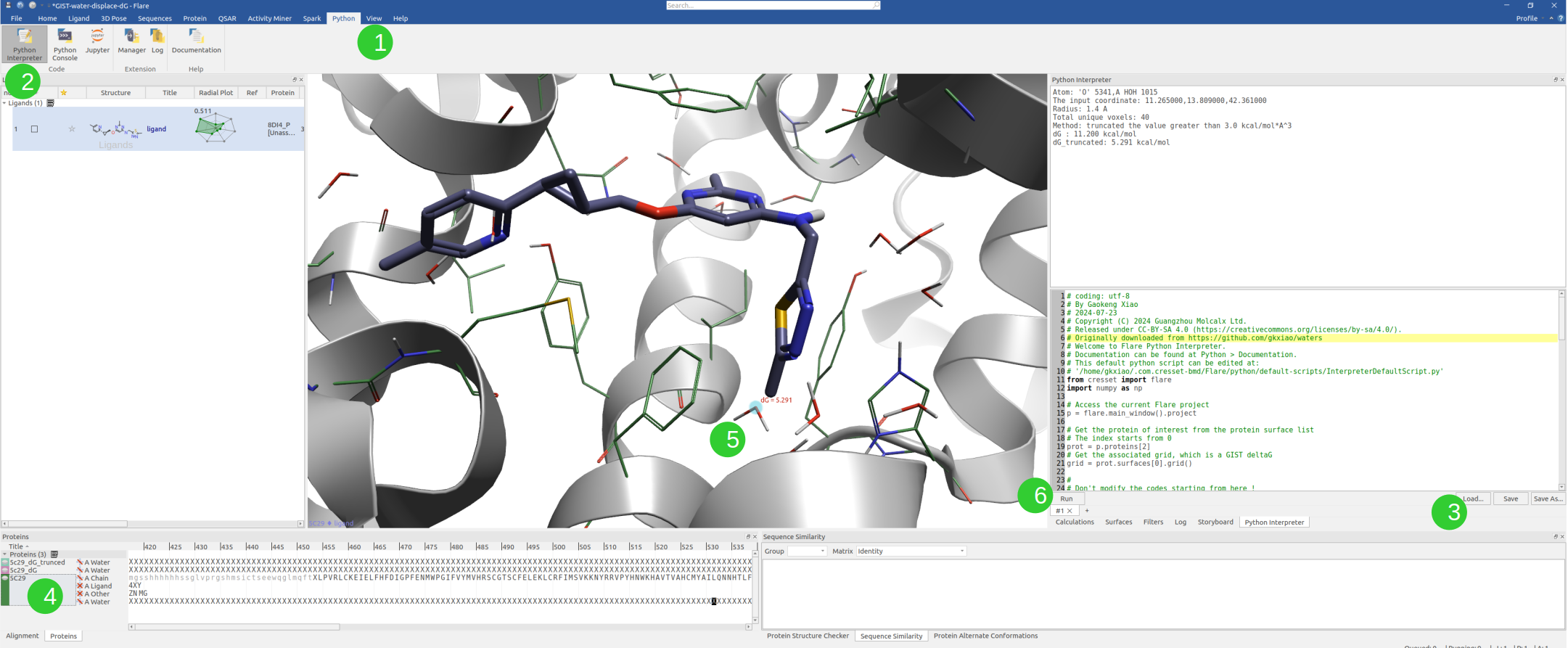
图19. 在Flare 3D视窗中呈现GIST ΔG操作步骤
文献
- Chen, J.M.; Xu, S.L.; Wawrzak, Z.; Basarab, G.S.; Jordan, D.B. Structure-based design of potent inhibitors of scytalone dehydratase: Displacement of a water molecule from the active site. Biochemistry 1998, 37, 17735–17744.
- Li, Z.; Lazaridis, T. Thermodynamics of buried water clusters at a protein-ligand binding interface. J. Phys. Chem. B 2006, 110, 1464–1475; Lazaridis, T. (1998) “Inhomogeneous fluid approach to solvation thermodynamics. 2. Applications to simple fluids,” Journal of Physical Chemistry B, 102(18). Available at: https://doi.org/10.1021/jp972358w.
- Haider, K.; Huggins, D.J. Combining solvent thermodynamic profiles with functionality maps of the Hsp90 binding site to predict the displacement of water molecules. J. Chem. Inf. Model. 2013, 53, 2571–2586.
- Robinson, D.; Bertrand, T.; Carry, J.-C.; Halley, F.; Karlsson, A.; Mathieu, M.; Minoux, H.; Perrin, M.-A.; Robert, B.; Schio, L.; et al. Differential Water Thermodynamics Determine PI3K-Beta/Delta Selectivity for Solvent-Exposed Ligand Modifications. J. Chem. Inf. Model. 2016, 56, 886–894.
- Ladbury, J.E.; Klebe, G.; Freire, E. Adding calorimetric data to decision making in lead discovery: A hot tip. Nat. Rev. Drug Discov. 2010, 9, 23–27.
- Barandun, L.J.; Ehrmann, F.R.; Zimmerli, D.; Immekus, F.; Giroud, M.; Grunenfelder, C.; Schweizer, W.B.; Bernet, B.; Betz, M.; Heine, A.; et al. Replacement of water molecules in a phosphate binding site by furanoside-appended lin-benzoguanine ligands of tRNA-guanine transglycosylase (TGT). Chem. A Eur. J. 2015, 21, 126–135.
- Biela, A.; Nasief, N.N.; Betz, M.; Heine, A.; Hangauer, D.; Klebe, G. Dissecting the hydrophobic effect on the molecular level: The role of water, enthalpy, and entropy in ligand binding to thermolysin. Angew. Chem. Int. Ed. 2013, 52, 1822–1828.
- Betz, M.; Wulsdorf, T.; Krimmer, S.G.; Klebe, G. Impact of surface water layers on protein-ligand binding: How well are experimental data reproduced by molecular dynamics simulations in a thermolysin test case? J. Chem. Inf. Model. 2016, 56, 223–233.
- Bucher, D., Stouten, P. and Triballeau, N. (2018) “Shedding Light on Important Waters for Drug Design: Simulations versus Grid-Based Methods,” Journal of Chemical Information and Modeling, 58(3), pp. 692–699. Available at: https://doi.org/10.1021/acs.jcim.7b00642.
- Graves, A.P. et al. (2017) “A Perspective on Water Site Prediction Methods for Structure Based Drug Design,” Current Topics in Medicinal Chemistry, 17(23). Available at: https://doi.org/10.2174/1568026617666170427095035.
- Nguyen, C.N., Kurtzman Young, T. and Gilson, M.K. (2012) “Grid inhomogeneous solvation theory: Hydration structure and thermodynamics of the miniature receptor cucurbit[7]uril,” Journal of Chemical Physics, 137(4), pp. 973–980. Available at: https://doi.org/10.1063/1.4733951.
- Flare™, version 9, Cresset®, Litlington, Cambridgeshire, UK; http://www.cresset-group.com/flare/; Cheeseright T., Mackey M., Rose S., Vinter, A.; Molecular Field Extrema as Descriptors of Biological Activity: Definition and Validation J. Chem. Inf. Model. 2006, 46 (2), 665-676; Bauer M. R., Mackey M. D.; Electrostatic Complementarity as a Fast and Effective Tool to Optimize Binding and Selectivity of Protein–Ligand Complexes J. Med. Chem. 2019, 62, 6, 3036-3050; Maximilian Kuhn, Stuart Firth-Clark, Paolo Tosco, Antonia S. J. S. Mey, Mark Mackey and Julien Michel Assessment of Binding Affinity via Alchemical Free-Energy Calculations J. Chem. Inf. Model. 2020, 60, 6, 3120–3130.
- Young, T. et al. (2010) “Dewetting transitions in protein cavities,” Proteins: Structure, Function and Bioinformatics, 78(8). Available at: https://doi.org/10.1002/prot.22699.
- Balius, T.E. et al. (2017) “Testing inhomogeneous solvation theory in structure-based ligand discovery,” Proceedings of the National Academy of Sciences, 114(33), pp. E6839–E6846. Available at: https://doi.org/10.1073/pnas.1703287114.
- Smith, C.R. et al. (2015) “Fragment-Based Discovery of a Small Molecule Inhibitor of Bruton’s Tyrosine Kinase,” Journal of Medicinal Chemistry, 58(14), pp. 5437–5444. Available at: https://doi.org/10.1021/acs.jmedchem.5b00734.
- SPARK替换结晶水分子. http://blog.molcalx.com.cn/2017/06/15/displacing-crystallographic-water-molecules-with-spark.html
- Young, T. et al. (2007) “Motifs for molecular recognition exploiting hydrophobic enclosure in protein–ligand binding,” Proceedings of the National Academy of Sciences, 104(3), pp. 808–813. Available at: https://doi.org/10.1073/pnas.0610202104.
- Uehara, S. and Tanaka, S. (2016) “AutoDock-GIST: Incorporating Thermodynamics of Active-Site Water into Scoring Function for Accurate Protein-Ligand Docking,” Molecules, 21(11), p. 1604. Available at: https://doi.org/10.3390/molecules21111604.
- Eberhardt, J. and Forli, S. (2023) “WaterKit: Thermodynamic Profiling of Protein Hydration Sites,” Journal of Chemical Theory and Computation, 19(9), pp. 2535–2556. Available at: https://doi.org/10.1021/acs.jctc.2c01087.
- PDE10A抑制剂MK-8189苗头到先导、先导优化中的水分子置换设计. http://blog.molcalx.com.cn/2024/05/17/lead-optimization-of-pde10a-inhibitor-mk-8189.html
- Hu, B. and Lill, M.A. (2012) “Protein pharmacophore selection using hydration-site analysis,” Journal of Chemical Information and Modeling, 52(4), pp. 1046–1060. Available at: https://doi.org/10.1021/ci200620h.
- Jung, S.W. et al. (2018) “Water Pharmacophore: Designing Ligands using Molecular Dynamics Simulations with Water,” Scientific Reports, 8(1), p. 10400. Available at: https://doi.org/10.1038/s41598-018-28546-z.
- Yoshida, S. et al. (2022) “Peptide-to-Small Molecule: A Pharmacophore-Guided Small Molecule Lead Generation Strategy from High-Affinity Macrocyclic Peptides.” Available at: https://doi.org/10.1021/acs.jmedchem.2c00919.
- 从肽到小分子——分而治之策略重现NNMT抑制剂的设计. http://blog.molcalx.com.cn/2023/04/10/from-peptide-to-small-molecule.html
- Wang, Y. et al. (2019) “Replacement of Protein Binding-Site Waters Contributes to Favorable Halogen Bond Interactions,” Journal of Chemical Information and Modeling, 59(7), pp. 3136–3143. Available at: https://doi.org/10.1021/acs.jcim.9b00128.
- David Becherer, J. et al. (2015) “Discovery of 4-Amino-8-quinoline Carboxamides as Novel, Submicromolar Inhibitors of NAD-Hydrolyzing Enzyme CD38,” Journal of Medicinal Chemistry, 58(17), pp. 7021–7056. Available at: https://doi.org/10.1021/acs.jmedchem.5b00992.
获取试用,请联系我们
想要在您自己的项目上尝试Flare GIST您可申请Flare试用版并了解如何将GIST用于其他基于结构设计的项目。


