
从肽到小分子——分而治之策略重现NNMT抑制剂的设计
摘要:本文以田野义小分子NNMT抑制剂的发现为例,联合使用Flare与Spark采用分而治之的策略重现了田野义从肽到小分子的发现过程,克服了大环肽类苗头分子的药效团尺寸过大而不能在一次虚拟筛选中命中化合物的问题,实现了从NNMT大环肽苗头化合物1到小分子抑制剂11的设计。
肖高铿/2023-04-05
前言
据报道1,2,大环肽能够以高亲和力和高选择性结合多种靶标,大环肽结合剂可靶向PPI(例如,K-Ras、HIV衣壳和TNF-α)、蛋白酶(例如, uPA、胰蛋白酶和HCV)以及其他多种靶标。虽然大环肽显示出广泛的应用性,但由于其主链酰胺结构,通常具有大分子尺寸和极性表面积;同时实现高亲和力和良好的类药特性仍然是一个挑战3。因此,如果可能,将大环肽结构跃迁为非肽小分子是克服其缺点的解决方案。
根据设计方法的差异,肽模拟物(Peptidomimetics)可分为 A、B、C、D等四个主要类别4。A类和B类模拟物具有肽样骨架,将原始肽的主链和/或侧链替换为非标准氨基酸或引入短链以提高活性5、细胞渗透性6、或抗蛋白水解降解性7。A类和B类仍然具有肽结构,并且肽的性质通常得以保留。C类和D类模拟物包括非肽类小分子骨架。C类,也称为蛋白质模拟物,已被开发用于抑制二级结构(如 α-螺旋和 β-折叠)介导的PPI,方法是用小分子骨架代替肽主链,同时保留侧链残基的方向和组成8。D类模拟物是一般的小分子,它们仅模拟生物活性肽的关键相互作用而没有直接的结构相似性。p53-MDM2相互作用抑制剂nutlins和PDK1变构抑制剂RS化合物的发现是文献报道为数不多的、成功的D类模拟物9,10。它们的发现是通过实验HTS方法来识别与每个表位肽具有低结构相似性的小分子骨架。尽管拟肽方法,尤其是D类,一直是识别小分子配体的一种有吸引力的方法,但大多数要模拟的目标肽仅限于PPI肽和天然表位肽。
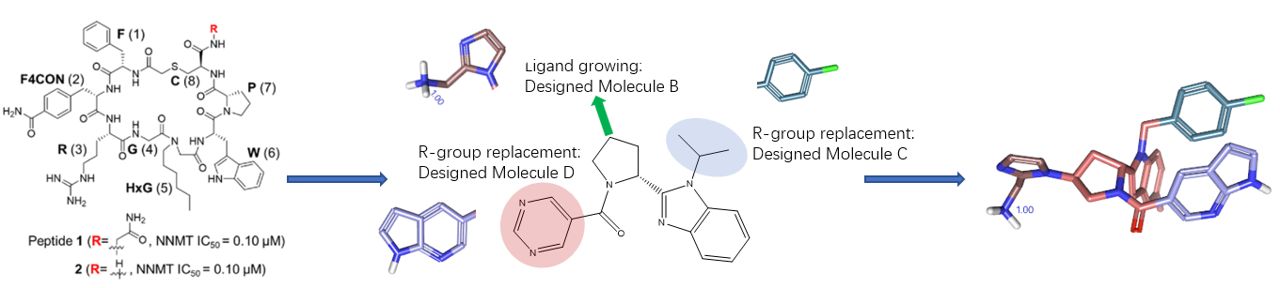
目前,人们已经通过展示肽筛选方法针对各种治疗靶标发现了很多大环肽配体11,12。然而,因为过大的分子尺寸和过高的极性表面积,这些肽类配体在平衡活性和诸如细胞渗透性等类药性上存在诸多挑战。为了克服这个问题,田野义制药公司的Yoshida等人13提出了一种新的从肽类到小分子先导的发现策略:结合大环肽展示筛选快速识别高亲和力肽类配体,然后用药效团指导从头小分子设计来克服肽类配体的成药性问题。
具体的讲,在Yoshida等人13的一项研究中,首先通过展示肽筛选技术鉴定了两个高亲和力NNMT大环肽配体1与2(图1),并通过结构生物学技术获得大环肽与NNMT的结合模式(PDB 7WMC)。
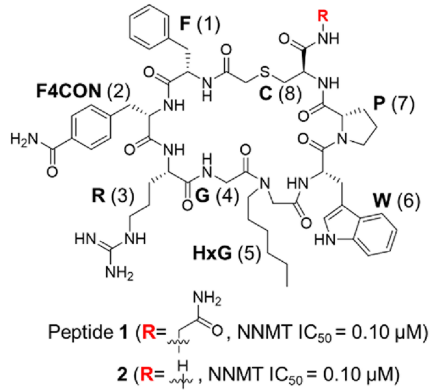
图1. NNMT大环肽配体1、2的化学结构
大环肽1与NNMT的结合模式如图2-a所示,大环肽1的Arg3-Trp6用洋红色棍表示、而环肽的其它残基用灰色线表示,蛋白NNMT用飘带表示。丙氨酸扫描实验表明Arg3-Trp6部分是大环肽1活性的关键部分。为了表征结合口袋的药效团,作者先用GIST方法14对结合位点进行了水合位点分析,结果如图2-b所示:从洋红色到绿色渐变的小球表示能量不利的水合位点,其中洋红色小球表示焓不利的水合位点,而绿色小球表示熵不利的水合位点。一方面,水的置换在配体结合中起到至关重要的作用,用与蛋白表面互补的基团置换能量不利的水是蛋白-配体结合的驱动力15。另一方面,水的能量学信息有助于确定抑制剂的潜在药效团16。在本次的结合位点水能量学分析阐明了大环肽1的Arg3、HxG5与Trp6的侧链在结合中所起的作用,并总结为如图2-d所示的药效团。药效团特征用彩色球体表示,疏水基团、氢键受体和带正电荷的基团分别用绿色、红色和蓝色球表示。
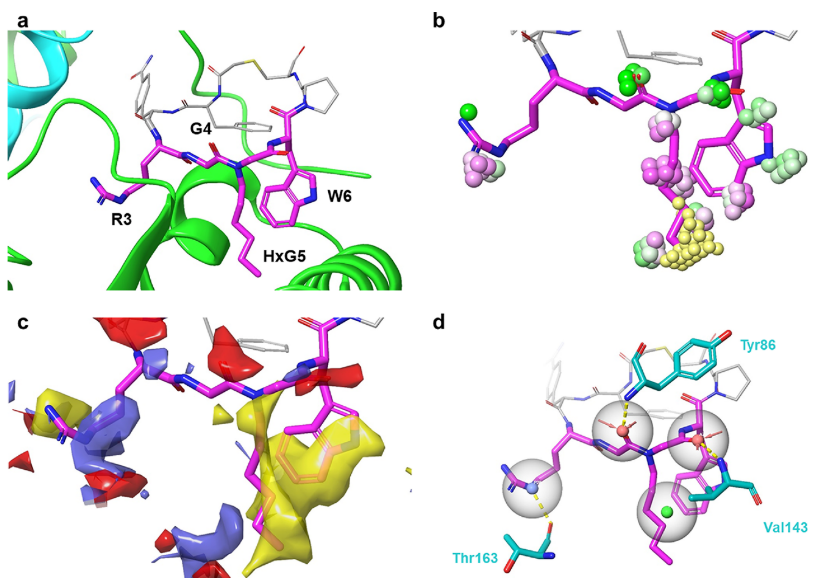
图2. 大环肽配体1的结合口袋分析
作者用分子对接对一个600万化合物库以及公司内部数据库进行了虚拟筛选,并用图2-d的药效团进行过滤,发现没有化合物可以满足全部的药效团特征。退而求其次,作者使用三个非常靠近的药效团特征(两个HBA和一个疏水特征)对对接结果进行过滤,发现Virtual Hit-A作为片段往同型半胱氨酸口袋延伸(图3 a,b)。接着以Virtual Hit-A为起点进行了计算结构优化。首先,为了满足同型半胱氨酸口袋中尚未匹配的药效团特征,设计了一个氨基通过适当刚性和合成可行性的连接臂伸向该口袋。其中,具有咪唑基亚甲基连接臂的分子-B能够能够以类似于大环肽1的方式与Thr163的主链羰基形成极性相互作用。然后,对苯并咪唑氮原子上的取代基进行了探索,设计了分子-C。在对接研究中,分子C的N-苄基苯并咪唑部分模拟了大环肽1的Trp6可以与Phe5侧链形成T形π-π相互作用。最后,对嘧啶片段进行了结构替换,发现氮杂吲哚基团可以与Asp142的侧链形成新的极性相互作用,并与Tyr86发生平行π-π堆积相互作用。在进一步计算优化后,设计了分子-D (11)可以模拟大环肽1的药效团(图3 c)。
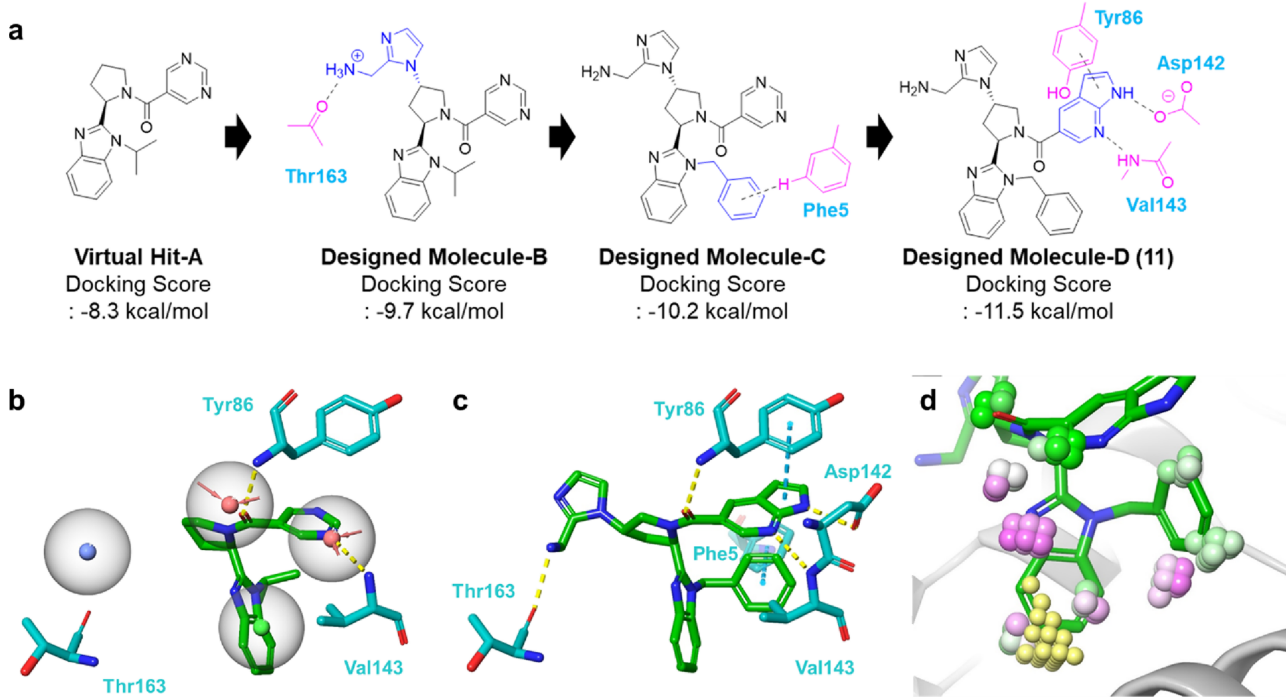
图3. 从虚拟筛选苗头化合物A(Virtual Hit-A)到抑制剂11的从头设计过程
对化合物11进行了合成与活性测试,IC50=54μM。对化合物11的进一步优化,发现了活性更强的系列化合物12-14,如图4所示。其中,还获得化合物13与NNMT的共晶结构(PDB 7WMT),进一步验证了整个设计过程假设的正确性。
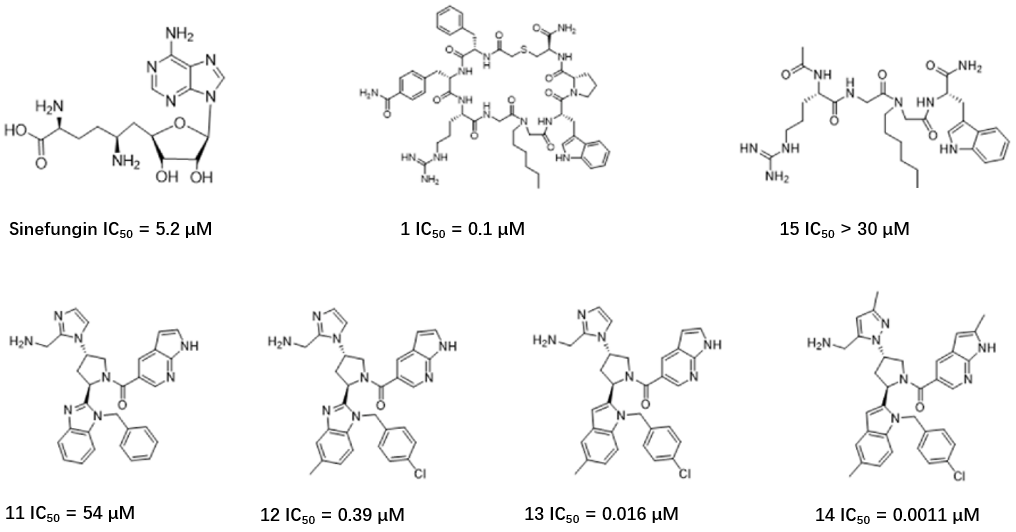
图4. NNMT抑制剂的SAR
本文的主要目的是,联合使用Flare与Spark,以大环肽1-NNMT共晶结构(PDB 7WMC)为基础,实现从Virtual Hit A出发到小分子化合物11的发现过程。实际上,这是个分而治之的策略:分别对各个子口袋进行设计直到拼合出最终化合物,如图5所示。
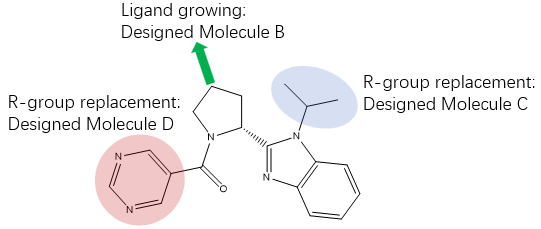
图5. 以VIrtual Hit A为起点分子的分而治之分子设计策略
结果与讨论
用分子对接预测Virtual Hit A的结合模式
Virtual Hit A是Peptide-to-small molecule的关键起点分子,其与NNMT的结合模式是用Flare Docking来预测,Virtual Hit A在大环肽1(Chain C)与NNMT的共晶结构(PDB 7WMC)结合位点里预测的结合模式如图6所示。

图6. 分子对接预测的VIrtual Hit A的结合模式
预测的结合模式(图6)与Yoshida等人13在图3-b描述的基本一致:苯并咪唑片段上的苯环与Ile62、Val143、Ala179以及Cys165发生疏水接触;苯并咪唑-N上的异丙基与Phe5以及Ala169发生疏水接触;苯并咪唑未被取代的N还与Cys165的巯基发生N-S相互作用;Virtual Hit A的四氢吡咯环与Cys165发生疏水接触;Virtual Hit A的羰基氧除了作为氢键受体与Tyr 86发生氢键相互作用之外,还与Cys141发生O-S相互作用;嘧啶环的一个氮作为氢键受体与Val 163的Cα上的酰胺NH发生氢键相互作用。
基于配体的分子生长:从Virtual Hit A到Designed Molecule-B
从Virtual Hit A到Designed Molecule-B的设计(图5绿色箭头)是通过Spark的分子生长策略,采用基于配体的打分来实现。Spark Query与约束如图7所示:以对接的Virtual Hit A的四氢吡咯上高亮的平伏键氢为连接点(图7-左),设置连接原子必须为环上的氮原子(可以是sp3或sp2杂化);并以PDB 7WMC的大环肽1(图7-右深绿色分子)为引导分子,往Arg3侧链(R3)方向生长,并设置末端胍基为正电中心的药效团约束(图7-右)。
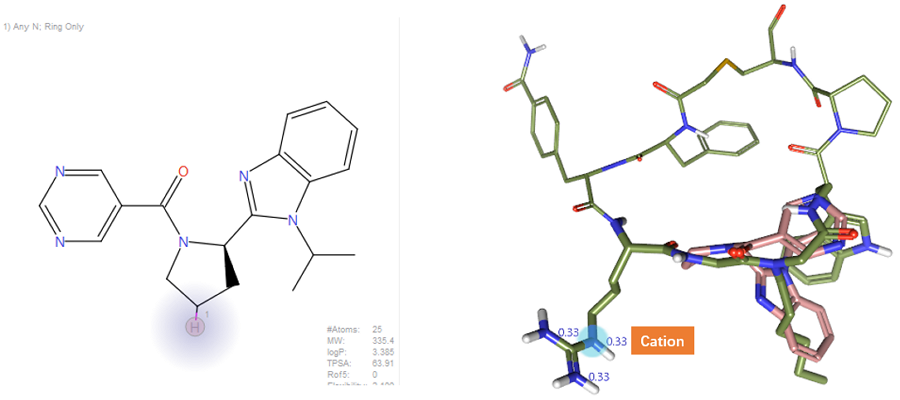
图7. 从Virtual Hit A到Designed Molecule-B的分子生长Spark query(左)与药效团约束(右)
计算的时候,使用了下面的设置:
- Calculation method: Normal
- Score method: Ligand Similarity (default)
- Databases to search: ChEMBL_common, Commercial Very Common与Common
这3个Spark片段数据库共总包含了95,568个片段可用于配体生长实验,其它参数用默认设置。

图8. 从Virtual Hit A到Designed Molecule-B几个代表性的分子生长结果
分子生长的部分结果如图8所示,其中黄色方框高亮的两个结果分别是Yoshida等人13报道的分子设计B(图3-a)与化合物14(图4)所用的杂环,除此之外,还有许多新颖的杂环。
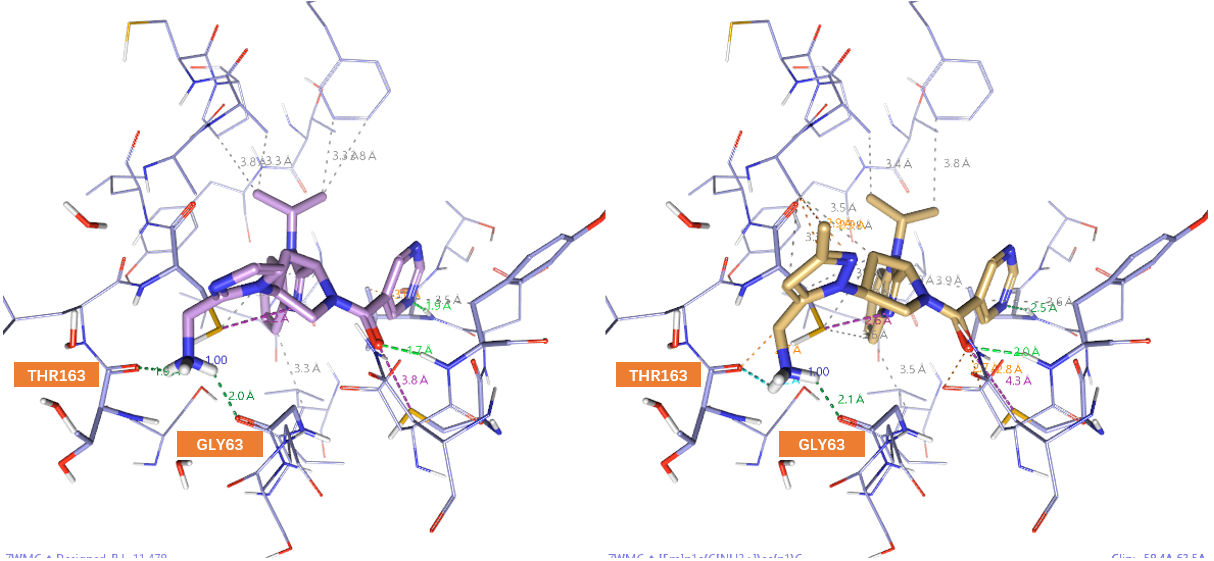
图9. 两个典型杂环片段的结合模式
如图9所示,两个典型侧链的结合模式满足我们的预期:末端质子化的阳离子胺基(图9)替换了大环肽1的阳离子胍基(图2-d)之后不仅与Thr163的羰基氧发生氢键相互作用,而且还与Gly63的羰基氧发生氢键相互作用。
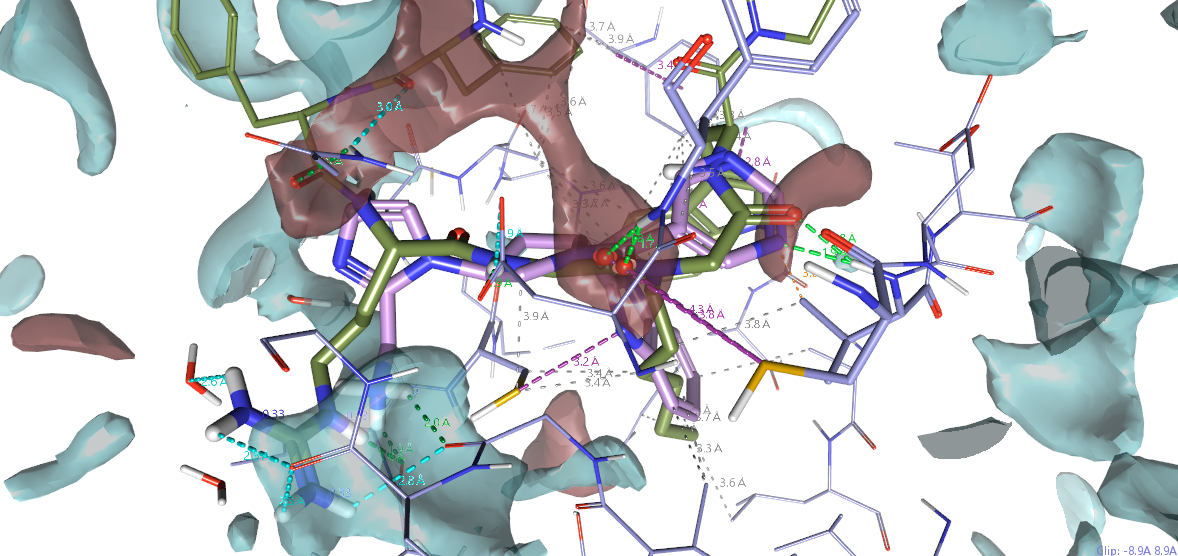
图10. NNMT同型半胱氨酸结合口袋的蛋白相互作用场(PIP)以及大环肽1(墨绿色)与分子设计B(粉色)的侧链比较
如图10所示,NNMT同型半胱氨酸结合口袋中,大环肽1(墨绿色)的胍基侧链与分子设计B(粉色)的质子化氨基侧链都被NNMT的正PIP相互作用场覆盖,这表明两者都与蛋白发生良好的静电互补。
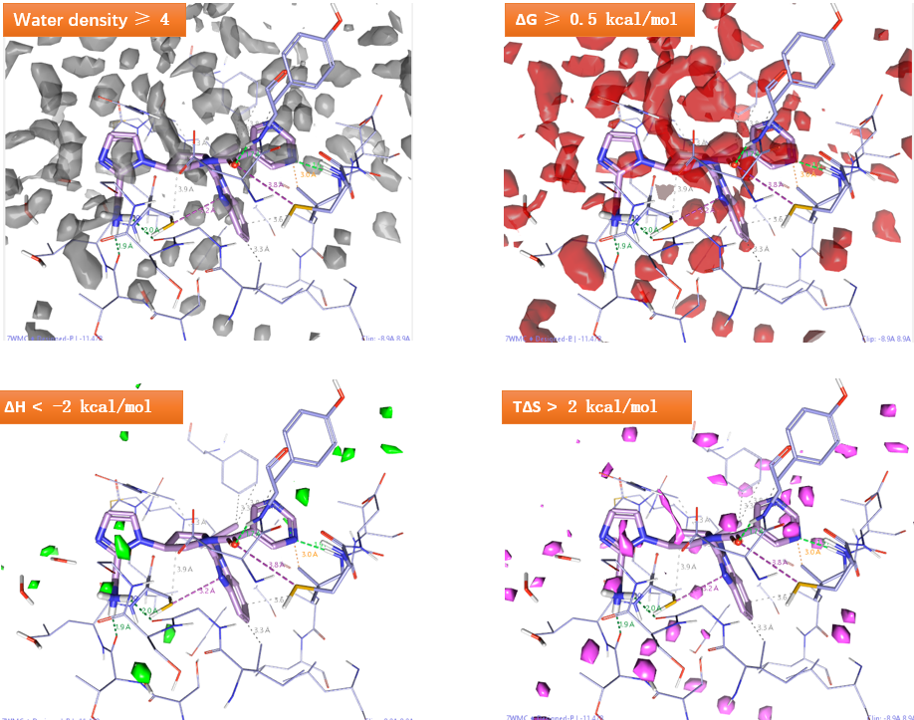
图11. NNMT(PDB 7WMC)大环肽1结合口袋的GIST分析
对NNMT的大环肽1结合位点的apo结构进行GIST分析,结果如图11所示,化合物B的质子化的氨基阳离子被一个高密度、高水合自由能(unhappy,ΔG大于1.50kcal/mol)的水覆盖,而且该处水的焓变为负值、熵变为正值。根据Ichihara等人的报道17,此类特征的水合位点宜引入与蛋白发生氢键相互作用的片段以便与蛋白表面发生互补的相互作用。
基于结构的基团替换:从Designed Molecule-B到Designed Molecule-C
以上一步分子生长获得化合物B为起点,进一步用基于结构的方法对苯并咪唑环N上的异丙基进行基团替换实验(图5蓝色高亮)。对异丙基进行基团替换的query如图12所示,为了让结果与文献尽量一致,本次实验设置连接点原子仅为sp2,sp3或C.ar类型的碳原子,这么做同时也兼顾了合成可行性,并同时设定对连接原子不限制是否为环原子。
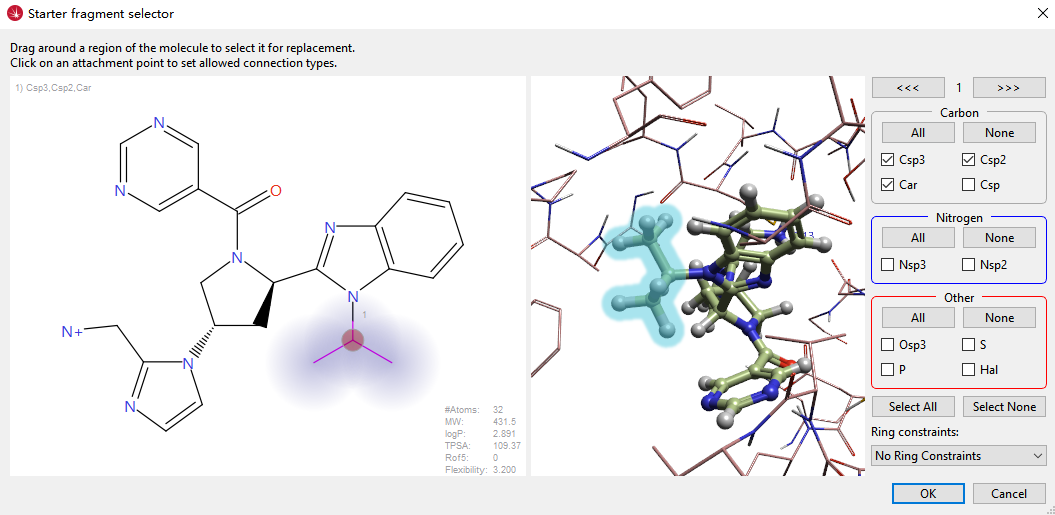
图12. 从Designed Molecule-B到Designed Molecule-C基团替换实验的query
基团替换用了如下参数设置:
- Calculation method: Normal
- Score method: Docking
- Filter | require: contains an aromatic ring
- Filter | exclude: contains a H-Bond donor; contains a H-Bond acceptor
- Databases to search: ChEMBL_common, Commercial Very Common与Common
其中Docking打分方法导致Spark对3个Spark片段数据库(共总包含了95,568个片段)进行基团替换实验时采用Lead Finder分子对接引擎进行打分;为了与该处的疏水药效团匹配,并设置过滤参数包含一个芳香环,不包含氢键受体或供体;其它参数用默认设置。
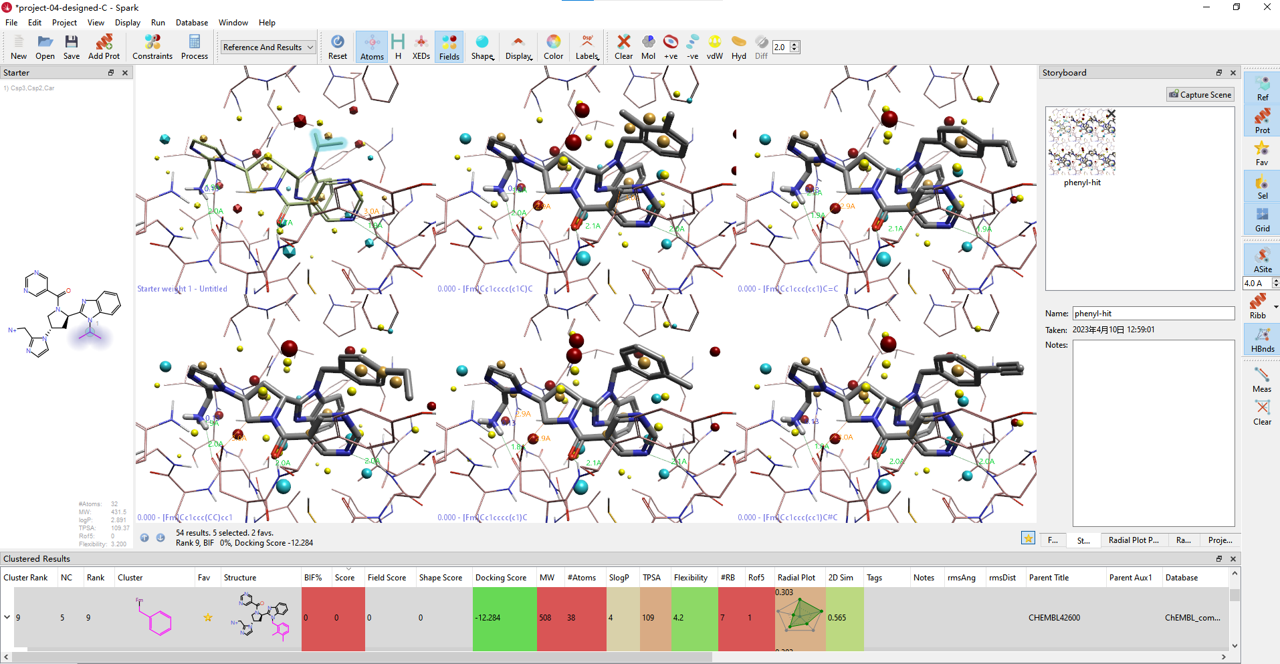
图13. 部分含苄基的结果
部分含苄基的结果如图13所示,这些苄基替换异丙基并与Phe5形成pi-pi堆积相互作用。根据GIST分析得到的热力学数据,可以进一步给出苄基位置的SAR与修饰建议,可以发现很多设计与GIST分析的结果互补匹配。
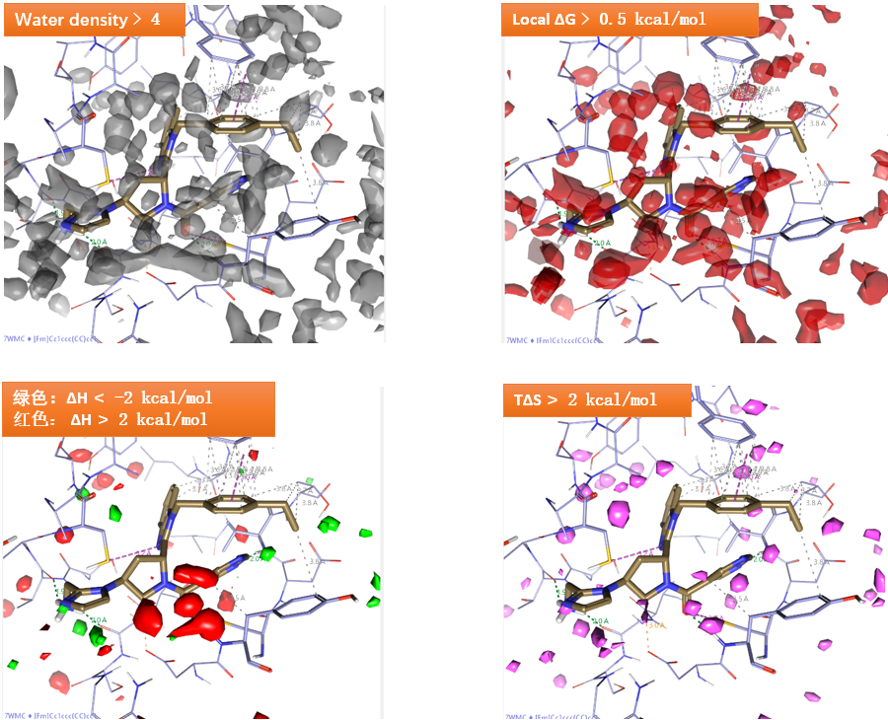
图14. 用GIST分析结果来解释Rank 17化合物的SAR
GIST对结合口袋的分析不仅可以用在Spark基团替换实验之前以提供对结合位点特征/SAR的理解,GIST的分析结果也可以用在Spark基团替换实验之后,用来发现更有潜力的化合物或为新化合物设计提供建议。以其中Rank 17化合物为例,GIST分析结果如图14所示,可以发现苄基片段与高密度(density大于4)、高自由能的水(ΔG大于0.5kcal/mol)重合,这说明从水合自由能上看,该子口袋是可被配体占据的。同时,苄基所在位置既没有ΔH小的,也没有ΔH大的区域,但是熵变相当大,因此该位置适合引入疏水基团。这些详细的水能量学信息进一步提示在苯基-4位引入氯等常见疏水基团来对高能水置换的方案,这可得到图4化合物12、13、14等设计。
基于配体的基团替换:从Designed Molecule-C到Designed Molecule-D(11)
以上一步获得化合物C的结合模式为起点,对图5红色高亮的嘧啶基团进行替换以引入更多的相互作用以实现结合亲和力优化。在基团替换的时候使用Docking作为打分函数可以利用蛋白信息从而引入新相互作用,比如上一步替换异丙基以及之前的案例18,19证明这是可行的。这个方法的前提是,有一个合适形状的蛋白结构口袋供分子对接使用。在这个算例中,我们发现由于口袋形状的原因,Spark Docking结果会错过目标化合物,需要用化合物13与NNMT共晶结构PDB 7WMT作为起点才能重现结果,但这不能反应真实的分子设计过程。因此,接下来用基于配体的打分,然后从结果里过滤出匹配药效团特征的结果。
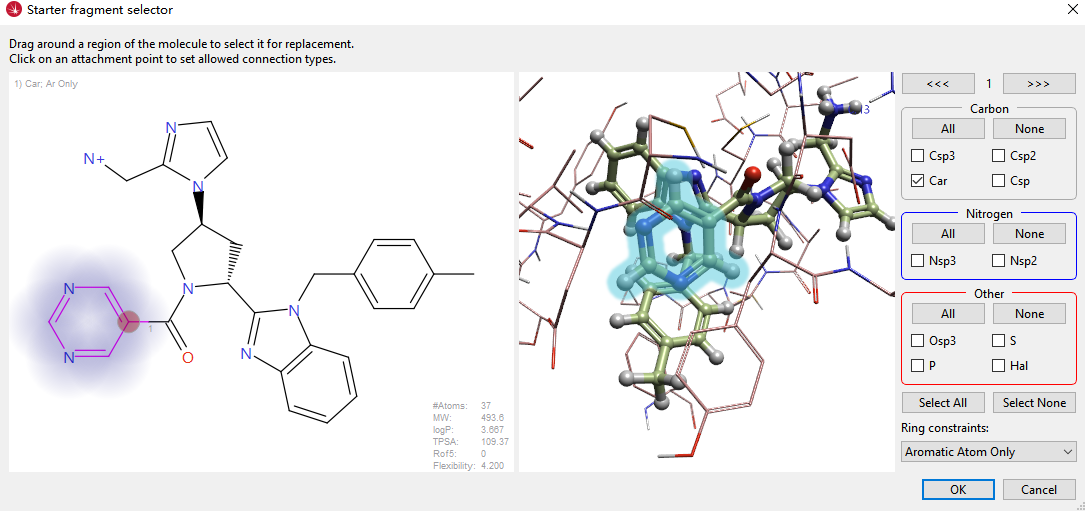
图15. 从Designed Molecule-C到Designed Molecule-D基团替换实验的query
从C到D的基团替换query如图15所示,为了让计算结果偏好于化合物D,设置连点原子类型为C.ar,并且限制为芳香环上的原子。Spark基团替换实验使用了下面的设置:
- Calculation method: Normal
- Score method: Ligand Similarity (default)
- Databases to search: ChEMBL_common
此外,还对Query设置了药效团约束,将与Val143酰胺NH发生氢键的嘧啶氮设置为氢键受体,如图16所示。其它参数用默认设置。ChEMBL_common数据库包含了41,681个片段,保留打分最高的500个分子作为输出结果。
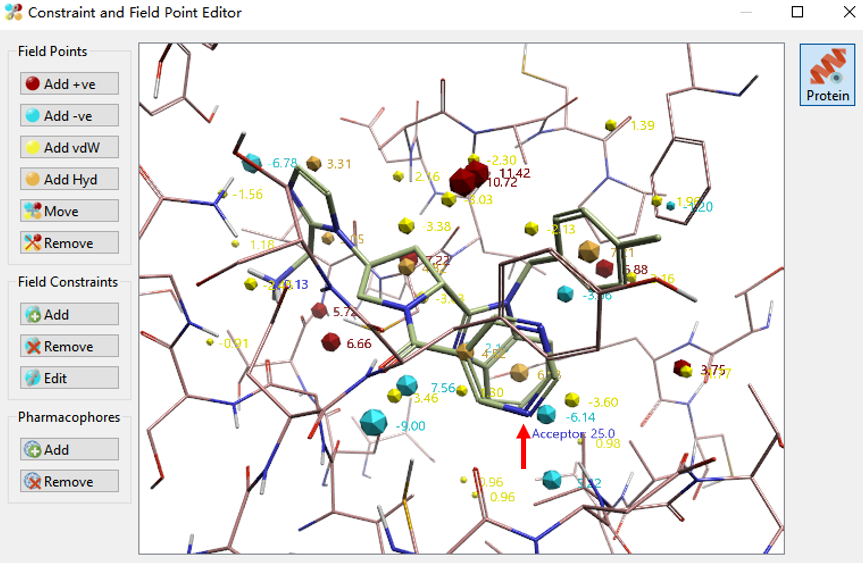
图16. 从Designed Molecule-C到Designed Molecule-D基团替换实验query的药效团约束
在命中的500个结果中,有113个化合物的BIF%大于60,其中包含打分排序为86的化合物D。如图17所示,你可以看到吡啶并吡咯的吡啶氮原子作为氢键受体与Val143发生氢键相互作用,而吡咯环NH作为氢键供体与Asp142的羧基氧原子发生氢键相互作用。总的来说,Rank 86化合物基于配体的打分相当高,Score=0.913, Field Score=0.893, Shape Score=0.933,而且引入了与Asp142的羧基氧原子新的相互作用。

图17. Spark Rank 86化合物为Designed Molecule-D
如图18所示,除了预期的吡啶并吡咯(图18-右)之外,Spark的基团替换设计中还包含其它令人感兴趣的新颖结果,比如图18-左与中所示的两个结构,均与吡啶并吡咯片段一样与Asp142、Val143发生相似的相互作用。

图18. 嘧啶环等排体替换的其它设计
小结
总的来说,从虚拟苗头化合物A出发,用Flare Docking预测结合模式,通过Spark基于配体或基于对接的打分对三个不同位置进行分子生长或取代基替换成功地重现了Yoshida等人13从大环肽1到小分子11系列化合物的设计。
方法
蛋白结构的准备
所有的蛋白结构都用Flare V6.121中的蛋白准备工具(Protein Preparation)来完成。下载结构并检查氨基酸侧链的替代构象,使用侧链的占据率或基于侧链的接触优选的侧链旋转异构体。这些操作是使用Flare的自动化蛋白准备工具来完成。该工作流的操作包含了:添加氢原子,探索可解离残基的互变异构体状态,添加缺失的侧链,并填补结构中两个氨基酸之间的缺口,末端残基的封端,优化残基侧链以遍最大化相互作用和内部氢键网络。在大多数情况下,Flare中的自动蛋白质准备适合于对接、能量计算与分子动力学模拟等研究,但仍建议对准备结果进行仔细地检查。
下载PDB 7WMC进行结构准备的时候,我们用C链的大环肽1来定义结合位点。在读入PDB结构之后,大环肽1的分子内C-S键是断裂的,需要手工用Protein Edit进行修复,然后将C链转化为配体,并提取出来。
GIST分析
Flare V6.1的3D-RISM与GIST用来完成这部分计算内容。对共晶结构PDB 7WMC的大环肽结合位点Apo结构的GIST分析采用3D-RISM预放置水分子再跑基于分子动力学模拟的GIST方式进行计算,详细过程参见原文20,主要参数如下:
- Calculation method: Normal
- Ligand: None
- Grid spacing:0.5 Å
- Grid Definition:Ligand
- Chains: A,B Chain, A 3DR
- Simulation length: 5 ns
- Solvent Model: explicit TIP4Pew Water
其中A 3DR为基于XED力场的3D-RISM预测的水分子链,Ligand为7WMC的C链大环肽1的Arg3-Trp6部分。
PIP分析
PIP分析采用Flare V6.1的Field Surface来完成,仅考虑准备好的7WMC的Chain A与B进行计算,不包含任何共晶水。
分子对接
分子对接采用Flare V6.1的Dock来完成,将准备好的7WMC的Chain A与B作为受体,用7WMC的C链大环肽1的Arg3-Trp6部分来定义grid,在Normal模式下进行分子对接计算。
分子生长与生物等排体替换
分子生长与生物等排体替换采用Spark V10.721实现,其中Spark数据库为2023年1月份版本。
结论
本文以田野义小分子NNMT抑制剂的发现为例,联合使用Flare与Spark采用分而治之的策略重现了田野义从肽到小分子的发现策略,克服了大环肽类苗头分子的药效团尺寸过大而不能在一次虚拟筛选中命中化合物的问题,实现了从NNMT大环肽苗头化合物1到小分子抑制剂11系列的设计过程。
文献
- Naylor, M. R.; Bockus, A. T.; Blanco, M. J.; Lokey, R. S. Cyclic Peptide Natural Products Chart the Frontier of Oral Bioavailability in the Pursuit of Undruggable Targets. Curr. Opin. Chem. Biol. 2017, 38, 141−147.
- Malde, A. K.; Hill, T. A.; Iyer, A.; Fairlie, D. P. Crystal Structures of Protein-Bound Cyclic Peptides. Chem. Rev. 2019, 119, 9861−9914.
- Vinogradov, A. A.; Yin, Y.; Suga, H. Macrocyclic Peptides as Drug Candidates: Recent Progress and Remaining Challenges. J. Am. Chem. Soc. 2019, 141, 4167−4181.
- Pelay-Gimeno, M.; Glas, A.; Koch, O.; Grossmann, T. N. Structure-Based Design of Inhibitors of Protein-Protein Interactions: Mimicking Peptide Binding Epitopes. Angew. Chem., Int. Ed. 2015, 54, 8896−8927.
- Sia, S. K.; Carr, P. A.; Cochran, A. G.; Malashkevich, V. N.; Kim, P. S. Short Constrained Peptides That Inhibit HIV-1 Entry. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 14664−14669.
- Lau, Y. H.; Andrade, P. D.; Quah, S. -T.; Rossmann, M.; Laraia, L.; Sköld, N.; Sum, T. J.; Rowling, P. J. E.; Joseph, T. L.; Verma, C.; Hyvönen, M.; Itzhaki, L. S.; Venkitaraman, A. R.; Brown, C. J.; Lane, D. P.; Spring, D. R. Functionalised Staple Linkages for Modulating the Cellular Activity of Stapled Peptides. Chem. Sci. 2014, 5, 1804−1809.
- Schafmeister, C. E.; Po, J.; Verdine, G. L. An All-Hydrocarbon Cross-Linking System for Enhancing the Helicity and Metabolic Stability of Peptides. J. Am. Chem. Soc. 2000, 122, 5891−5892.
- Horne, W. S.; Grossmann, T. N. Proteomimetics as Protein- Inspired Scaffolds with Defined Tertiary Folding Patterns. Nat. Chem. 2020, 12, 331−337.
- Vassilev, L. T.; Vu, B. T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; Fotouhi, N.; Liu, E. A. In Vivo Activation ofthe p53 Pathway by Small-Molecule Antagonists of MDM2. Science 2004, 303, 844−848.
- Rettenmaier, T. J.; Sadowsky, J. D.; Thomsen, N. D.; Chen, S. C.; Doak, A. K.; Arkin, M. R.; Wells, J. A. A Small-Molecule Mimic of a Peptide Docking Motif Inhibits the Protein Kinase PDK1. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 18590−18595.
- Sohrabi, C.; Foster, A.; Tavassoli, A. Methods for Generating and Screening Libraries of Genetically Encoded Cyclic Peptides in Drug Discovery. Nat. Rev. Chem. 2020, 4, 90−101.
- Wang, X. S.; Chen, P. -H. C.; Hampton, J. T.; Tharp, J. M.; Reed,C. A.; Das, S. K.; Wang, D. -S.; Hayatshahi, H. S.; Shen, Y.; Liu, J.; Liu, W. R. A Genetically Encoded, Phage-Displayed Cyclic-Peptide Library. Angew. Chem., Int. Ed. 2019, 58, 15904−15909.
- Yoshida, S.; Uehara, S.; Kondo, N.; Takahashi, Y.; Yamamoto, S.; Kameda, A.; Kawagoe, S.; Inoue, N.; Yamada, M.; Yoshimura, N.; et al. Peptide-to-Small Molecule: A Pharmacophore-Guided Small Molecule Lead Generation Strategy from High-Affinity Macrocyclic Peptides. 2022. https://doi.org/10.1021/acs.jmedchem.2c00919.
- Nguyen, C. N.; Young, T. K.; Gilson, M. K. Grid Inhomogeneous Solvation Theory: Hydration Structure and Thermodynamics of the Miniature Receptor Cucurbit[7]uril. J. Chem. Phys. 2012, 137, 973− 980.
- Wang, L.; Berne, B. J.; Friesner, R. A. Ligand Binding to Protein- Binding Pockets with Wet and Dry Regions. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 1326−1330.
- Jung, S. W.; Kim, M.; Ramsey, S.; Kurtzman, T.; Cho, A. E. Water Pharmacophore: Designing Ligands Using Molecular Dynamics Simulations with Water. Sci. Rep. 2018, 8 (1), 10400. https://doi.org/10.1038/s41598-018-28546-z.
- Ichihara, O.; Shimada, Y.; Yoshidome, D. The Importance of Hydration Thermodynamics in Fragment-to-Lead Optimization. ChemMedChem 2014, 9 (12), 2708–2717. https://doi.org/10.1002/cmdc.201402207.
- 肖高铿. KRAS抑制剂MRTX849骨架跃迁引入新的相互作用. 墨灵格的博客. http://blog.molcalx.com.cn/2021/04/26/mrtx849-scaffold-hopping.html
- 从活性位点里发现新的蛋白-配体相互作用. 墨灵格的博客. http://blog.molcalx.com.cn/2021/01/23/spark-docking.html
- 肖高铿. 联合使用3D-RISM与GIST计算密闭结合位点里水的热力学性质. 墨灵格的博客. http://blog.molcalx.com.cn/2022/07/18/3d-rism-and-gist.html
- Flare V6.1. https://www.cresset-group.com/software/flare
- Spark V10.7. https://www.cresset-group.com/software/spark


