
摘要:本文深入分析了Flare两种轻量级LF VSscore与EC score打分方法对ACK1激酶抑制剂铰链结合片段优先级排序的性能,旨在为骨架跃迁提供高效、准确的方法。Flare的EC score作为一种快速的筛选工具,成功揭示了抑制剂与ACK1激酶结合位点间的静电相互作用对活性的影响,其与实验数据的显著线性关系验证了静电差异是活性差异的主要原因。相互作用成分更完整的SQM ΔEinter打分进一步提高了对不同母核的排序能力,表现出与实验值的高相关性。Flare的LF VSscore虽然在预测精度上存在一定的误差,但其在活性与非活性化合物的区分上表现出色,为药物发现的早期阶段提供了有效的富集手段。总的来说,Flare的LF VSscore与EC score提高了筛选效率,加速了DMTA运转而加速药物的发现。
作者:肖高铿/2024-07-26
前言
激活型Cdc42相关酪氨酸激酶1 (Activated Cdc42-associated tyrosine Kinase 1,ACK1)是非受体型酪氨酸激酶,一个有潜力的抗癌治疗药开发的靶标。Jiao等人1报道了6个ACK1激酶抑制剂,它们具有不同化学类型的铰链区结合片段且活性跨度大(4.4 kcal/mol),结构与ACK1的Ki如图1所示。
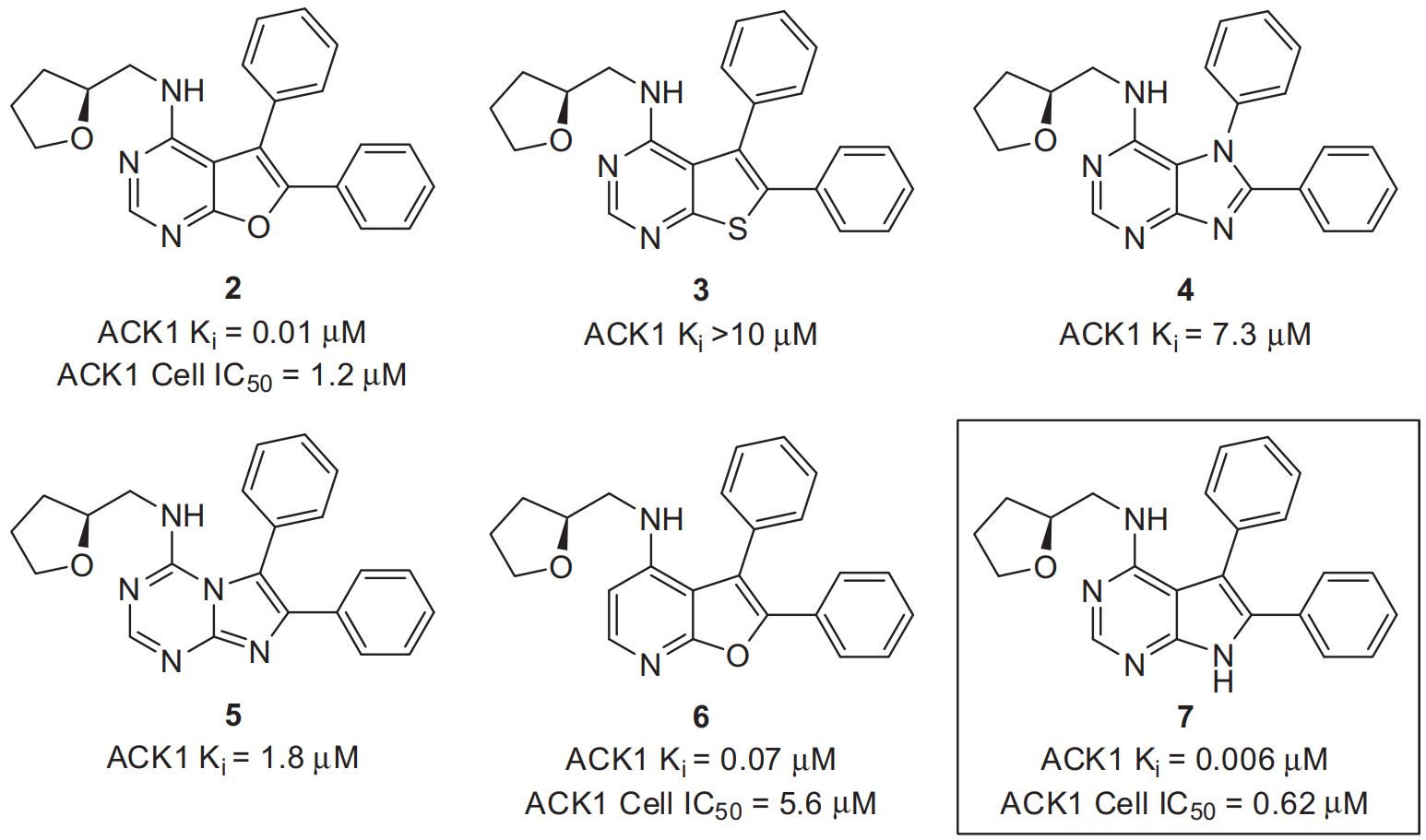
图1. 6个具有不同铰链结合片段的ACK1抑制剂及其Ki值,图片来自文献[1]
先采用快速的方法对大量的化合物进行计算,然后再用基于MD的MM/GBSA、FEP等精确、但计算速度慢、也更昂贵的方法对其中一部分进行过滤是目前虚拟筛选常用的方法。然而,根据Granadino-Roldán等人的报道2,用炼金术FEP结合自由能计算方法对该系列SAR进行优先级排序是有挑战的,主要因为计算结果受到蛋白结构准备的影响。
表1. 五个不同结合自由能计算方法的性能2
| Protocol | Pearson R2 | MUE | Slope |
|---|---|---|---|
| A | 0.36±0.07 | 1.55±0.06 | 0.3 |
| B | 0.67±0.03 | 1.29±0.03 | 0.4 |
| C | 0.5±0.2 | 1.53±0.07 | 0.2 |
| D | 0.84±0.03 | 1.29±0.03 | 0.5 |
| E | 0.33±0.08 | 1.73±0.08 | 0.3 |
MUE: Mean Unsigned Error。
在这个研究中,作者用了5个方法准备蛋白结构:A)用分子对接预测结合模式,接着用MM/PBSA打分取打分最佳的结合模式用于FEP计算;B)手动选择结合模式用于FEP计算;C)在A的基础上,手动在口袋关键位置放置结合水以反映对该体系知识的理解,然后用于FEP计算;D)在B的基础上,手动在口袋关键位置放置结合水以反映对该体系知识的理解,然后用于FEP计算;E)在A的基础上,每个λ窗口模拟时长延长至10倍。结果如表1所示,用docking结合MM/PBSA打分选择的结合模式用于结合自由能计算的方法(A、B、C)比人工选择结合模式的方法(B、D)预测性能差,这说明计算结果受起始结合模式的影响很大,即使延长模式时间至10倍(方法E)也无法改善预测性能。方法C比A以及方法D比B的线性相关性系数地到显著提升,这说明蛋白结构深处的关键水非常影响结果。其中方法D兼顾了结合模式选择与关键水处理,表项出最佳的预测性能,MUE=1.29 ± 0.03 kcal/mol,Pearson R2=0.84。
表2. Uni-FEP计算的与实验的结合自由能比较2
| Compound | ΔGExp(kcal/mol) | ΔGUni-FEP(kcal/mol) | ΔGUni-FEP_H2O(kcal/mol) |
|---|---|---|---|
| 2 | -10.91 | -9.82±0.20 | -9.92±0.06 |
| 3 | -6.82 | -9.54±0.78 | -8.44±0.15 |
| 4 | -7.01 | -7.47±0.23 | -7.68±0.050 |
| 5 | -7.84 | -9.12±0.22 | -9.05±0.99 |
| 6 | -9.76 | -8.30±0.29 | -8.50±0.10 |
| 7 | -11.22 | -9.31±0.29 | -9.97±0.07 |
注:ΔGUni-FEP为未加水分子的计算结果,ΔGUni-FEP_H₂O为加水分子后的计算结果。
用Uni-FEP对同一个数据集的研究结果(见表2)进一步证实了关键水的处理对计算结果至关重要3。当蛋白体系中不含有水分子时,计算的ΔGUni-FEP与实验ΔG误差较大(RMSE=1.64 kcal/mol),计算值与实验值之间没有线性关系,Pearson R2=0.16。然后根据Granadino-Roldán等人2对口袋深处水分子的描述,手动放置水分子之后再计算的ΔGUni-FEP_H₂O,不仅误差变小(RMSE=1.20),而且计算值与实验值之间具有显著地线性关系,Pearson R2=0.681。加水之后的FEP计算可以把活性较高的分子排序靠前,如图2所示,比如化合物2与7得到正确排序,聚集在左下角。虽然如此,但是还有高、低活性化合物未得以区分。活性高的6在纵坐标上(预测值)显著地高于5而与活性最低的3相当,这说明ΔGUni-FEP_H₂O尚未能将高活性的6与低活性的3、5区分开。
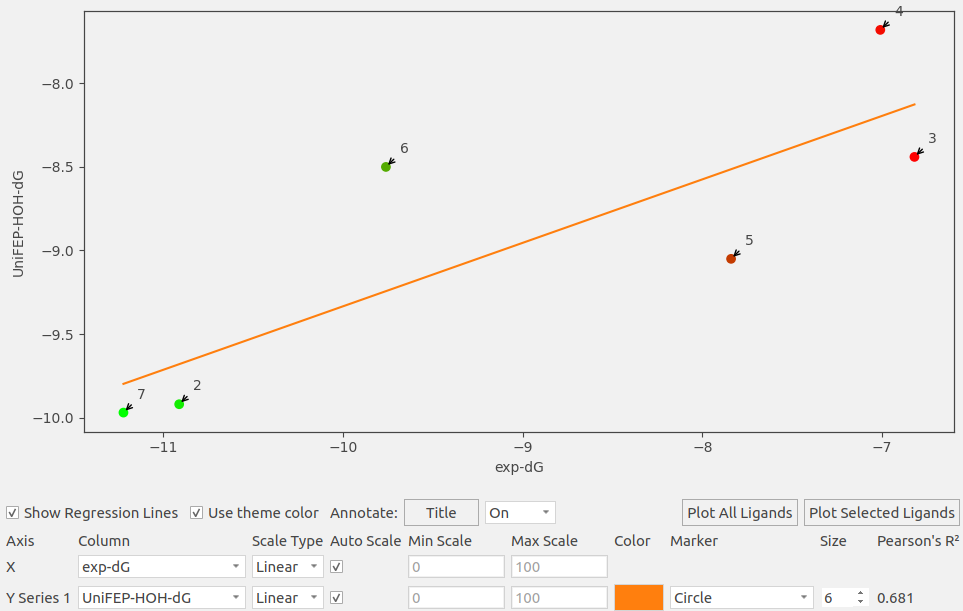 图2. 预测值ΔGUni-FEP_H₂O与实验值Exp. ΔG的比较,Exp. ΔG从低到高对应从亮绿色到鲜红色。
图2. 预测值ΔGUni-FEP_H₂O与实验值Exp. ΔG的比较,Exp. ΔG从低到高对应从亮绿色到鲜红色。
此外,在未加水Uni-FEP的计算实验中,仅一个原子差异的分子对2-3没被计算方法区分开,这反应了另一个问题:力场可能还有缺陷,不能区分五员杂环噻吩与呋喃。如果去掉化合物3,则Pearson R2从0.16提高到0.51。
在药物发现漏斗(级联过滤)中,FEP方法作为理论最严格、高计算量的结合自由能计算方法通常处于最后步骤对前期挑选的化合物进行排序。然而,如前所述的ACK1抑制剂的FEP计算,无论是Granadino等人2还是Uni-FEP3的案例都表明,计算结果与蛋白结构的准备非常相关。在第一轮“开箱即用”模式下FEP对化合物排序性能很差,在知道答案的情况下,可以改进流程提高预测性能,这也说明了FEP计算在正式Production模式之前跑一个Benchmark模式的重要性。
因为XED力场与SQM可以克服力场对硫原子描述不准确的问题,所以本文的主要目的之一是以这6个ACK1抑制剂骨架跃迁数据集为例,用Flare的XED力场与SQM计算抑制剂与ACK1之间的相互作用能,并评估其对活性的排序能力。本文的另一个目的是演示Flare基于结构的轻量级计算用于优先级排序的性能,在这个例子中,可达到文献FEP方法相似或更好的性能,在不知道正确“答案”的情况下可为后续重型计算(比如FEP)提供最大的保障。
结果
用Electrostatic ComplementarityTM打分进行排序
静电互补性打分(Electrostatic Complementarity score,EC score)是一个已经充分验证的方法4,其基本思想是:当配体和受体的静电势匹配(即具有相同的值和相反的符号)时,实现配体和受体之间的最大静电亲和力。之前,我们成功地用Flare内置的XED力场计算的静电相互作用与静电互补性(EC)分析对不同化学类型的TYK2 JH1铰链结合片段进行的优先级排序5。鉴于这些EC打分的成功案例以及其速快速、高效的特性,我们决定先尝试使用EC对ACK1抑制剂进行分析。选择EC打分的另一原因是,在这个ACK1抑制剂数据集中的配体都紧密相关,它们的大小近乎相同,具有大致相同数量的可旋转键,具有一致形状与结合模式,静电差异很可能是它们主要的活性差异原因。
以Jiao等人1解释ACK1共晶结构PDB 4EWH中的抑制剂35为模板,用Flare Conf Hunt & Align将化合物2-7叠合到共晶的抑制剂上,结果发现化合物2-7全局最低能构象与4EWH共晶配体叠合,与铰链区相互作用模式如图3所示。使用分子叠合而不是完全自动的分子对接,是因为叠合可以得到噪音最小化的结合模式,就像生成一个好的3D-QSAR时一样。
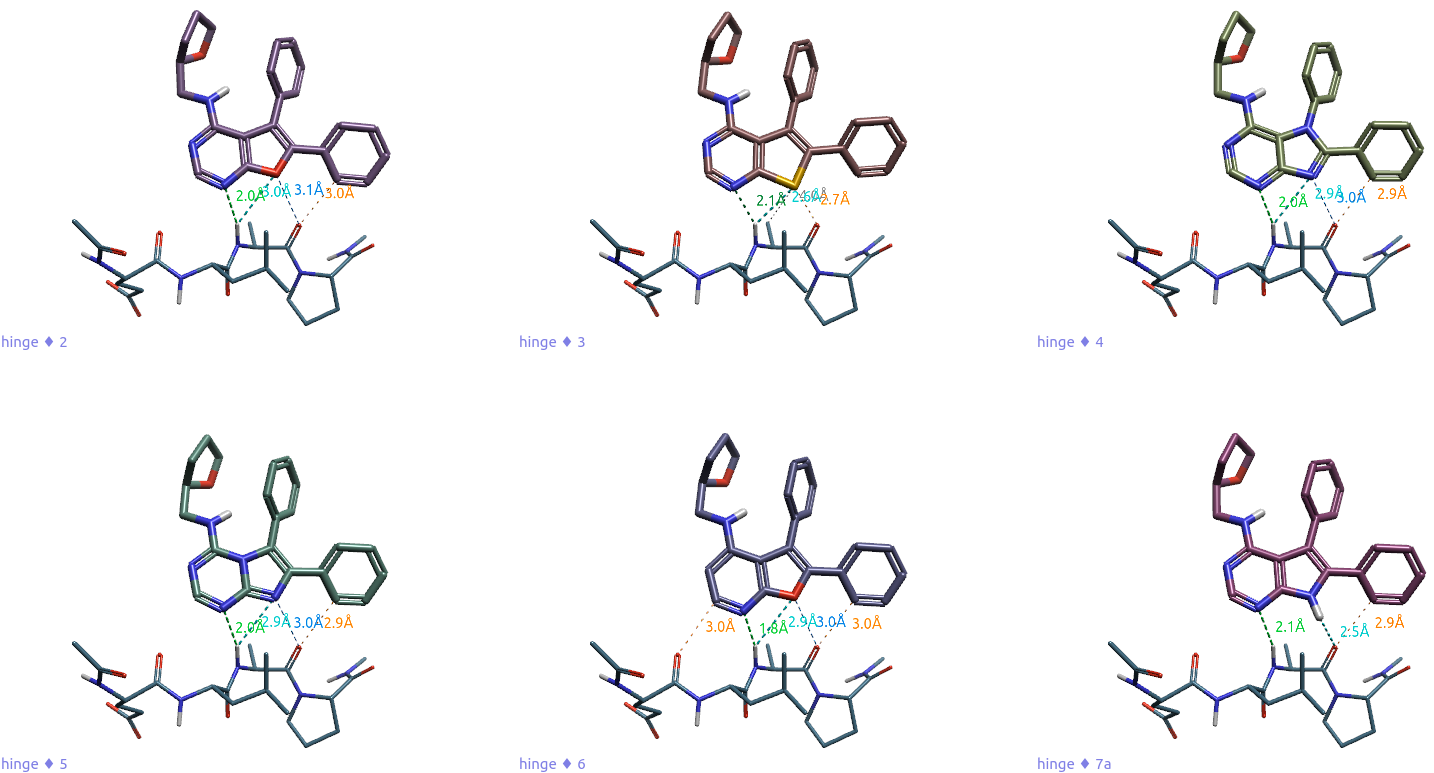
图3. ACK1铰链区与6个抑制剂的结合模式
用Flare EC score对6个抑制剂在蛋白“干”的结合位点里进行静电互补性打分,结果如图4所示,发现EC score与结合自由能之间具有显著的线性关系,Pearson R2=0.45,这说明静电互补性差异确实是它们活性差异的主要原因。同时也发现,化合物2、3在横坐标(实验值)距离很远(ΔΔG=4kcal/mol),而在纵坐标上非常靠近,这说明作为预测器的EC打分对区分2、3的能力不够好。高活性(绿色)的6、7与低活性(红色)的4、5分别位于纵坐标的两端,说明EC对这4个化合物分类正确。
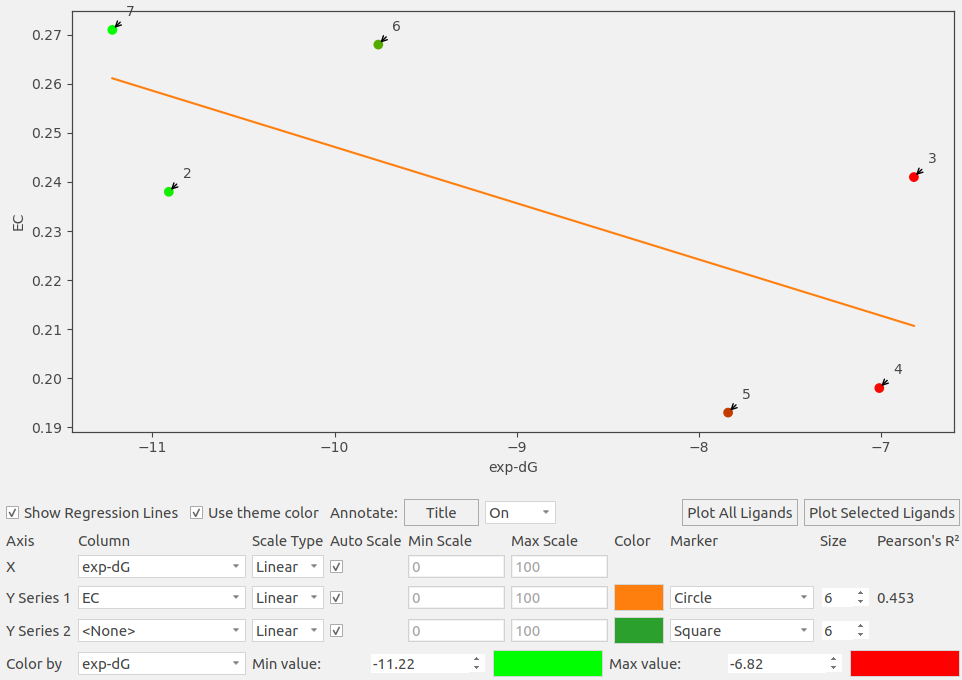
图4. 抑制剂-ACK1的静电互补性打分(EC)与实验结合自由能(Exp. ΔG)之间关系
与Uni-FEP相比,EC打分虽然具有较低的Pearson相关性系数,但是在活性与非活性的分类能力上两者相当,甚至EC略占优势。考虑到单因素的EC打分取得良好的效果,且6个化合物主要差异在于与铰链区相互作用部分,这促使我们尝试使用量子力学方法(Quantum Mechanics,QM)来计算配体与蛋白之间的相互作用能,有可能通过完整的相互作用分析而取得更好的效果。
用SQM相互作用能(ΔEinter)进行排序
使用QM计算方法有可能弥补EC单因素相互作用的不足,因此我们决定使用半经验量子力学(Semiempirical Quantum Mechanics,SQM)程序xTB6来计算蛋白与配体间的相互作用能(ΔEinter):
$$
ΔE_{inter} = E_{complex} – E_{hinge} – E_{ligand} \cdots (1)
$$
其中Ecomplex、Ehinge与Eligand分别表示在GFN2-xTB理论水平6计算的配体-铰链区复合物、铰链区、配体的能量。用铰链区的5个残基Thr205-Glu206-Leu207-Ala208-Pro209-Leu210来带替蛋白,用Flare 从PDB 4EWH提取出来,并进行结构准备,封端。铰链区进一步用QM在PB86-D3BJ/DEF2-SVP进行两面角约束的几何优化,配体在PB86-D3BJ/DEF2-TZVP进行几何优化,然后在GFN2-xTB理论水平进行单点能计算,并最后计算相互作用能,结果如表3所示。
表3. 抑制剂-铰链区残基的SQM ΔEinter、实验的与预测的结合自由能比较
| ID | Ecomplex | Ehinge | Eligand | ΔEinter(kcal/mol) | Exp. ΔG(kcal/mol) | Pred. ΔG(kcal/mol) |
|---|---|---|---|---|---|---|
| 2 | -185.7957 | -108.5659 | -76.9007 | -206.5358 | -10.91 | -9.780 |
| 3 | -184.9626 | -108.5659 | -76.0729 | -203.2210 | -6.82 | -6.532 |
| 4 | -185.4452 | -108.5659 | -76.5523 | -205.1769 | -7.01 | -8.449 |
| 5 | -185.4480 | -108.5659 | -76.5567 | -204.1745 | -7.84 | -7.466 |
| 6 | -185.5124 | -108.5659 | -76.6169 | -206.8282 | -9.76 | -10.067 |
| 7 | -185.1734 | -108.5659 | -76.2760 | -208.0331 | -11.22 | -11.248 |
注:如果没有说明,单位为a.u.。
在SQM ΔEinter与实验结合自由能(Exp. ΔG)之间存在统计学意义上显著的线性相关性,Pearson R2=0.81(p-value = 0.014),如下图5所示。
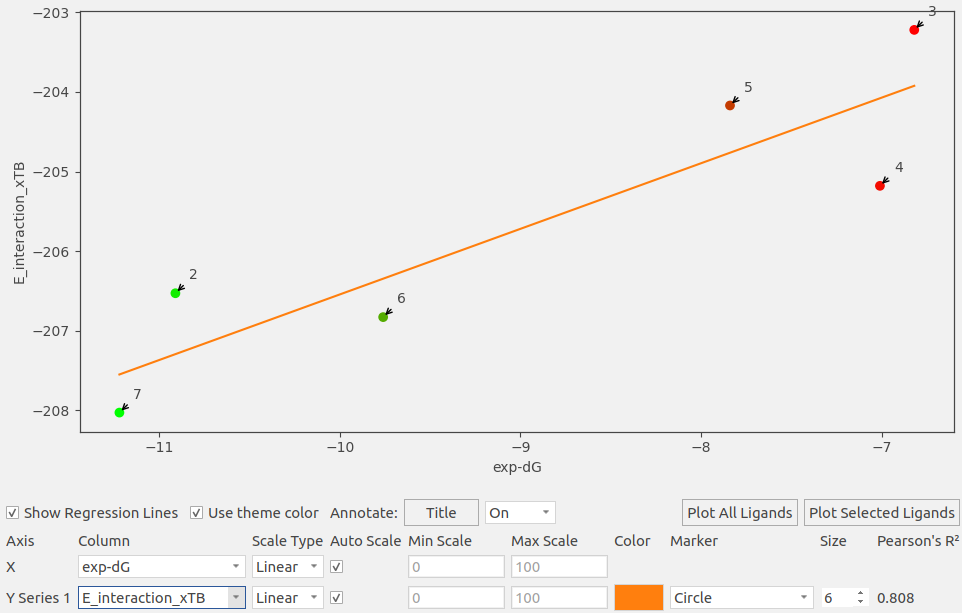
图5. SQM ΔEinter(纵坐标)与实验结合自由能(Exp. ΔG,横坐标)散点图
SQM ΔEinter是结合自由能的强预测器,在Flare Python界面里用scipy软件包的stats模块拟合得到线性回归方程(2):
$$
Pred. ΔG = 0.9801 \times ΔE_{inter} + 192.6453 \cdots (2)
$$
根据方程(2)预测的结合自由能(Pred. ΔG,见表1)与实验的结合自由能之间的均方根误差相当小,RMSE=0.78。其中,化合物4的预测误差最大,为1.44 kcal/mol,其它化合物的预测误差均小于0.5 kcal/mol。SQM ΔEinter可用于合成实验前的优先级排序,如图5所示,三个活性强(绿色)的与三个活性弱(红色)的化合物被完美地分为两组。虽然如此,有两对化合物在各自的亚组内排序错误。在三个绿色的强活性组里,化合物6比2的实验ΔG低了1.15 kcal/mol,但计算值却高了0.29 kcal/mol;在三个红色低活性组里,化合物4比5的实验ΔG高了0.83 kcal/mol,但计算值却低了0.78 kcal/mol。这说明,虽然与铰链区的相互作用差异是这几个化合物活性差异的主要原因,但是还有其它因素影响活性,这需要与FEP等其它方法一起联合使用,取长补短。
SQM ΔEinter排序的成功并非偶然。Pecina等人7的研究表明,使用半经验QM相互作用能作为打分函数的SQM score 2.20在PL-REX数据集上优于分子对接打分,与实验数据具有优秀的相关性,平均Pearson R2 = 0.69,重要的是,在全部靶标上取得一致的优秀性能。本研究中的SQM ΔEinter与Pecina的SQM 2.20打分差异在于没有考虑:1)溶剂化效应;2)构象熵;3)构象张力能惩罚。虽然如此,但是化合物2-7具有非常相似的结构,它们溶剂化与构象效应对结合自由能的影响差异很小,并且预测的结合构象都是低能构象,构象张力能惩罚小到可忽略不计。
用分子对接打分进行排序
如图1所示,化合物7铰链结合片段上的五员环吡咯比另外5个化合物的五员环多出一个氢键供体,化合物7还与PDB 4EWH共晶配体具有同样的铰链结合片段,这提示,可能另外5个化合物的结合模式与化合物7稍有不同。为此,制定了使用不同蛋白结构的分子对接打分策略:
- 化合物7:采用ACK1的共晶结构PDB 4EWH进行对接
- 化合物2-6:采用分子动力学模拟的构象进行对接
化合物7与PDB 4EWH的共晶配体35具有完全相同的铰链结合片段以及相似的侧链,因此假设化合物7与共晶配体可能具有相似的结合模式,直接用共晶结构PDB 4EWH对7进行分子对接打分。
以活性较强化合物2为代表,将前述2的结合模式为起点进行10ns的分子动力学模拟,取MD产物阶段(production)平衡后的一帧蛋白构象、优化后用于分子对接打分。
在进行分子对接打分之前有必要先了解一下PDB 4EWH结合口袋的特点。观察Fo-Fc=3δ的电子密度差图,如图6所示,可以发现在配体嘧啶并吡咯环的嘧啶N原子附近有一个绿色电子密度差区块,这意味这里有原子没有被解释出来,合理的猜测是有水分子未被解释。
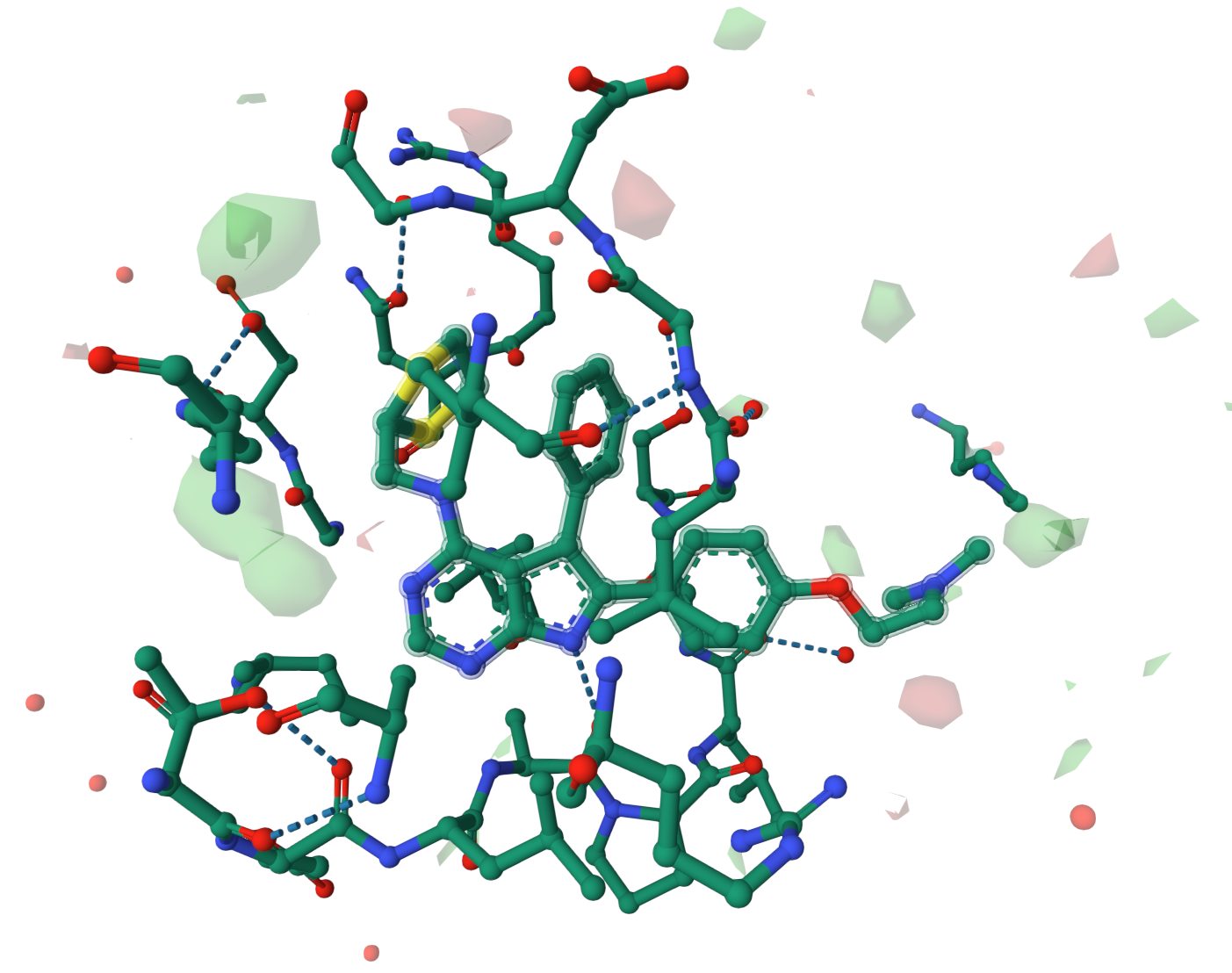
图6. PDB 4EWH共晶配体周围fo-fc电子密度差图。绿色:+3δ,红色:-3δ
为了验证这个猜测,用Flare对PDB 4EWH的结合口袋进行了GCNCMC水增强采样的GIST-apo分析,结果如图7所示,在靠近配体嘧啶N原子附近Fo-Fc=3δ的正值区确实被高密度的水覆盖(图7左),并且为Local Unhappy water(ΔG值大于0.5)等值图覆盖,为可替换的水。注意:这是对apo结合口袋计算,有可能结合不同配体之后,该处的水可能变成happy water。到此,Fo-Fc=3δ正值区缺失水的假设可以被确认,并且与不同的配体结合,可能继续保留,也可能被替换。这为后续的Flare docking打分时将靠近嘧啶N原子的水设置为flickering提供了强有力的支持,最终让Flare docking在打分时根据配体特征自行决定该水分子的去、留。
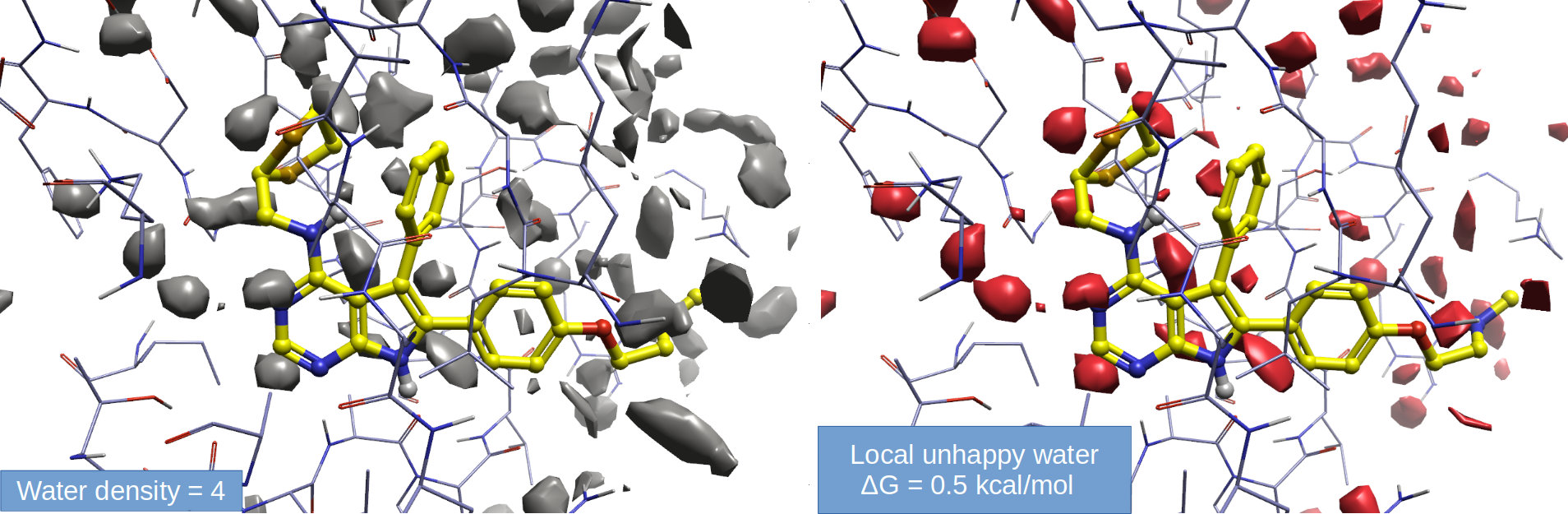
图7. PDB 4EWH结合口袋GIST-apo分析结果。左:水密度等值图;右:Local Unhappy water等值图。
现在,用Flare Dynamics对化合物2/PDB 4EWH复合物进行GCNCMC水分子增强采样的5ns分子动力学模拟,以MD产品阶段的一帧快照(第4969帧)来说明体系稳定时的结合位点特征。
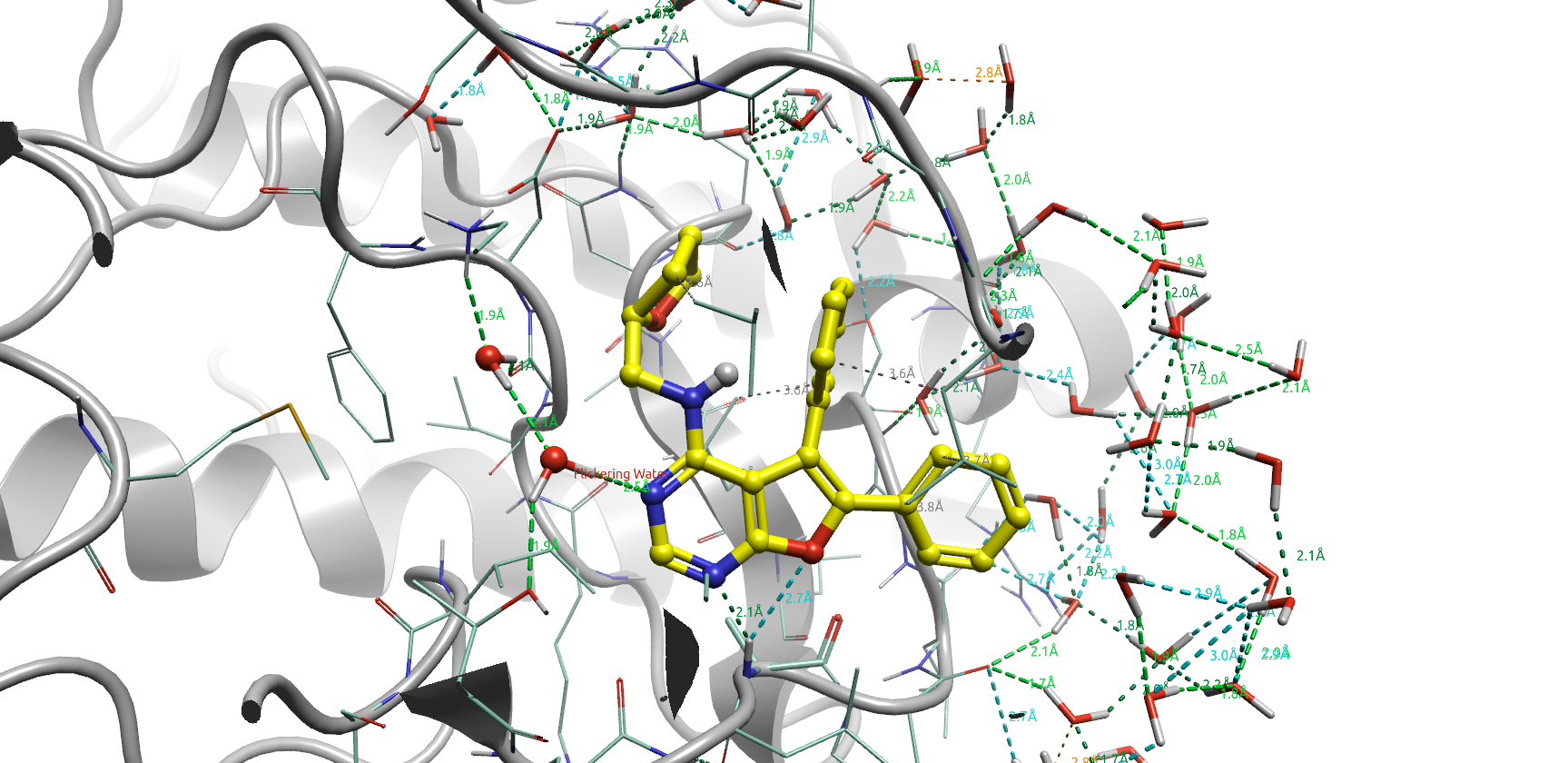
图8. 化合物2/ACK1复合物分子动力学模拟的一帧快照(第4969帧)
如图8所示,在化合物2的嘧啶氮原子附近可以发现两个水分子,它们出现在Fo-Fc=3δ正值区(图6嘧啶附近的绿色区块),并通过氢键网络,介导了蛋白与配体之间的相互作用。这说明分子动力学模拟采集到了被晶体学家们忽略的水。在Flare里对该快照进行标准的蛋白结构准备,几何优化,作为ACK1抑制剂2-6分子对接打分的蛋白结构使用。
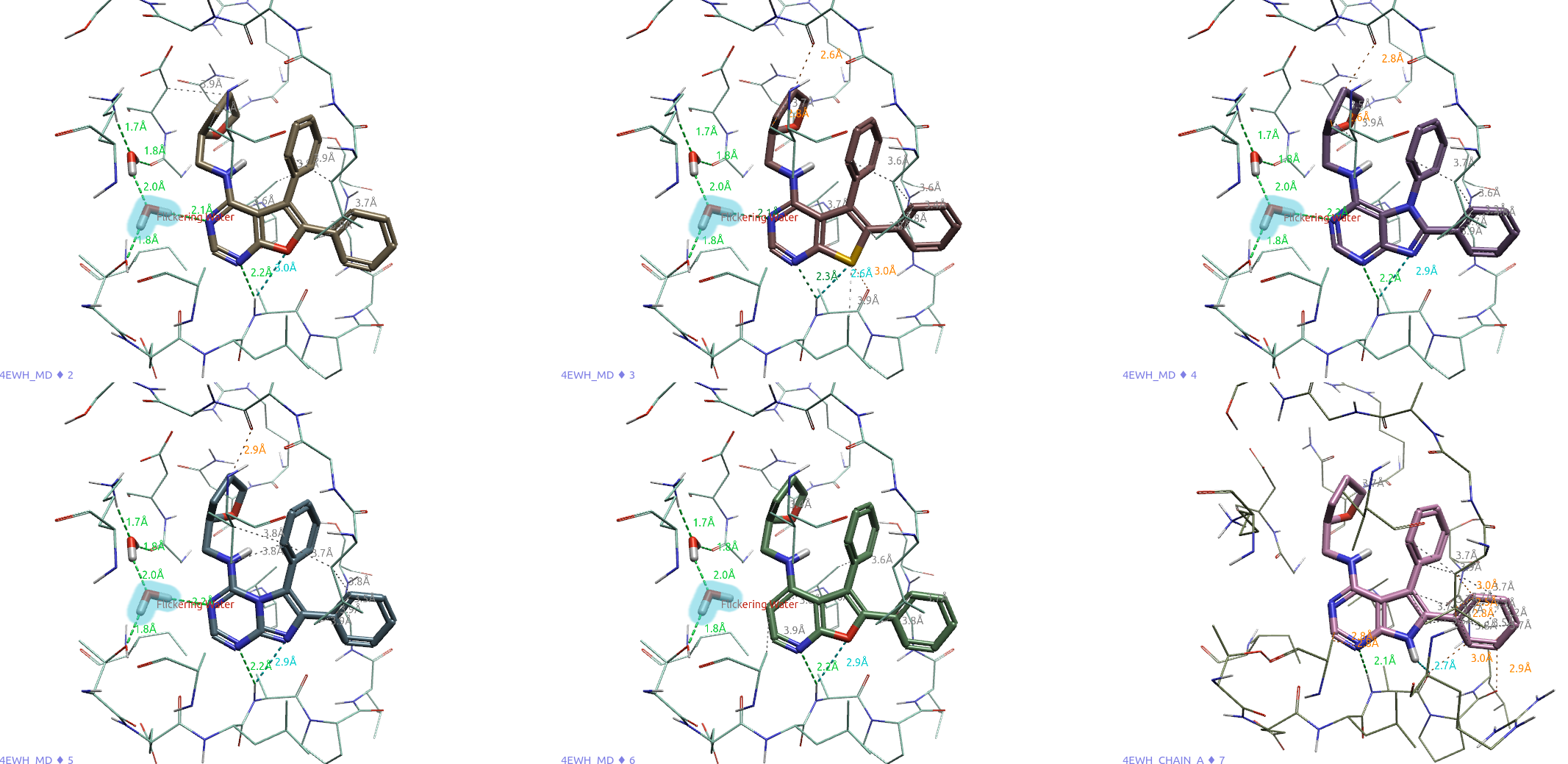
图9. 抑制剂2-7在ACK1结合口袋里进行分子对接打分,高亮的那个水被设置为flickering water,让对接打分函数自己决定它的去留。
Flare Docking分子对接打分结果表明,抑制剂2-6在ACK1结合口袋里打分时,都保留了flickering water。抑制剂7直接在PDB 4EWH的结合口袋里打分。Flare Docking的计算引擎为Lead Finder,它使用LF VSscore来对虚拟筛选的不同化合物进行打分、排序。最终6个化合物的LF VSscore打分值与实验结合自由能见表4。
表4. 抑制剂2-7的Flare Docking打分结果与实验结合自由能
| Compound | LF VSscore (kcal/mol) | Exp. ΔG(kcal/mol) |
|---|---|---|
| 2 | -12.277 | -10.91 |
| 3 | -10.261 | -6.82 |
| 4 | -9.335 | -7.01 |
| 5 | -9.721 | -7.84 |
| 6 | -12.511 | -9.76 |
| 7 | -13.015 | -11.22 |
可以发现,如图10所示,分子对接打分函数LF VSscore的打分值与实验值具有极其显著的线性相关性,Pearson R2=0.868(p-value=0.007)。更重要的是,LF VSscore正确将6个化合物分为跨度足够大的两组(图10-纵坐标),并与实验值(图10-横坐标)一致,完美地将高活性与低活性区分开。
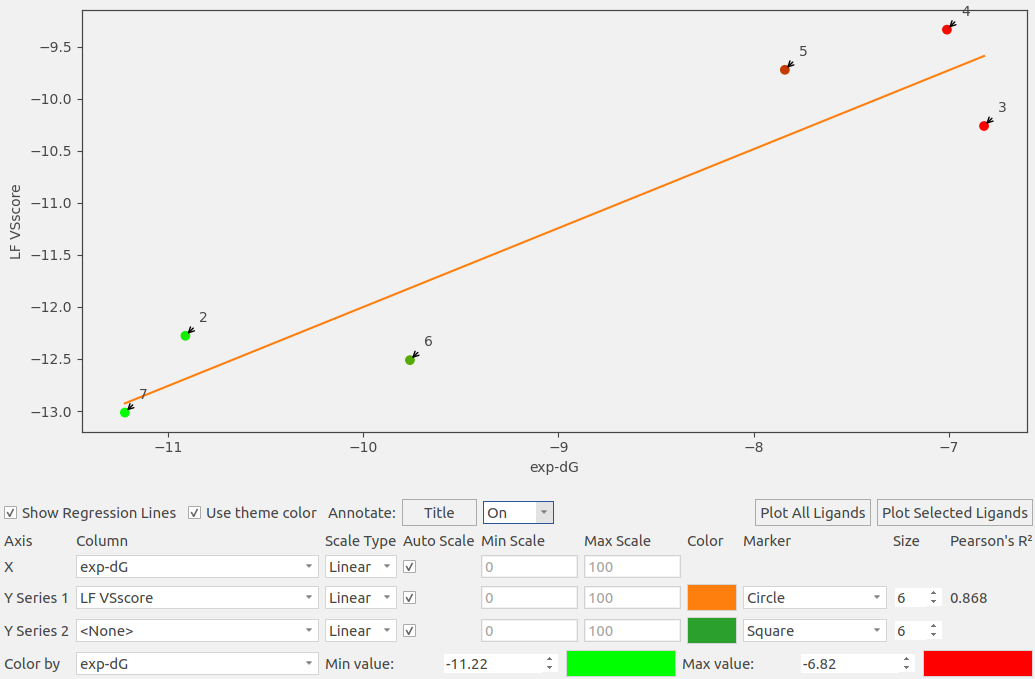
图10. 抑制剂2-7在ACK1结合口袋里的分子对接打分LF VS-score与实验结合自由能Exp. ΔG具有显著的线性关系,Pearson R2=0.868
LF VSscore与Exp. ΔG虽然具有显著的相关性,但是它们之间的RMSE=2.36,MAE=2.26,这个误差并不适合于在先导化合物优化中用来进行优先级排序。LF VSscore具有良好的区分活性与非活性分子的能力使得它更适合于在虚拟筛选中用来初步富集活性化合物。
用Flare轻量级计算加速药物发现
在设计-制造-测试-分析(Design-Make-Test-Analyze,DMTA,图11)方法中会大量用到最先进的科学技术,并强调速度的重要性。这意味着开发人员,在面对药物发现过程中的多变量优化,不仅要达到一定的准确度,而且需要速度与通量,以便项目尽可能多次地绕DMTA运转起来以加速药物发现。
图11. DMTA模型
在这过程中,我们所能提供的是分级计算方法,这些方法随着计算成本的增加而变得更加准确,通常人们称之为计算级联(cascade of computational method)或发现漏斗(Drug discovery funnel,图12)。漏斗的上半部分是轻量级的计算方法,通常是筛选和过滤化合物;而下半部分是重量级的计算方法,通常是预测结合自由能等定量计算。
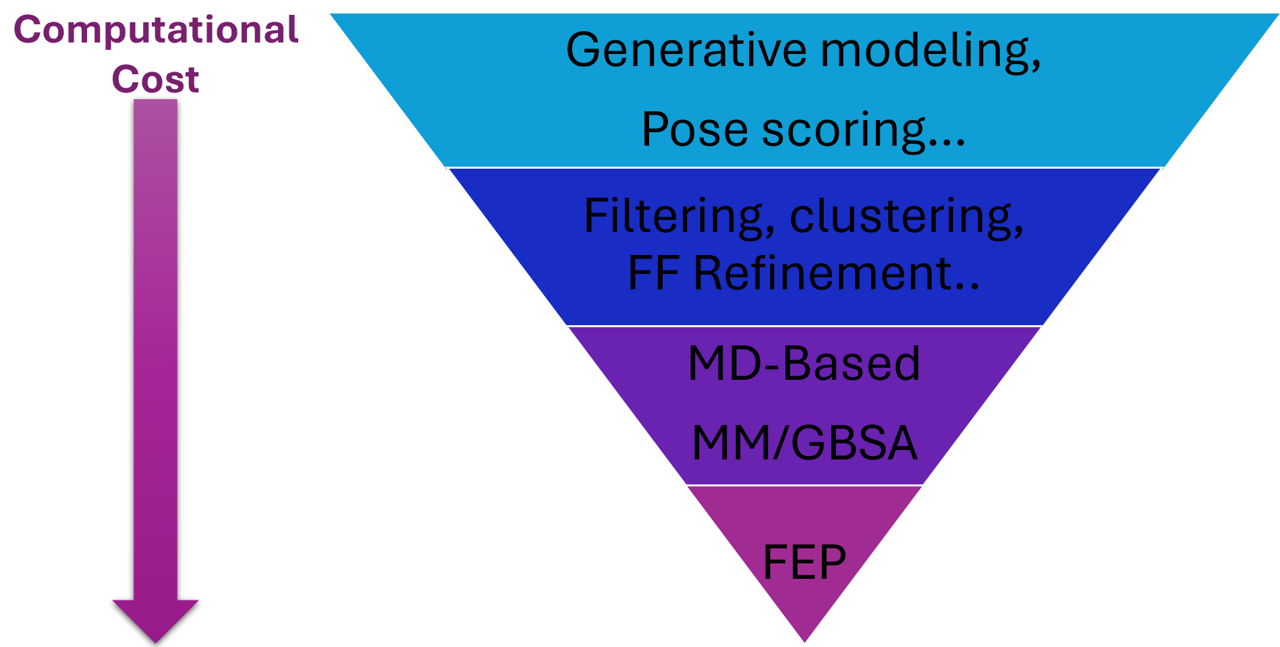
图12. 药物发现漏斗
显然,在漏斗中任一阶段都很重要。在本文中,我们展示了Flare两个轻量的基于结构的计算方法EC score与LF VSscore对不同骨架ACK1抑制剂的优先级排序性能,以及对EC进行拓展的SQM ΔE score的性能。
在本研究中,ACK1数据集是一系列仅在铰链结合片段上有差异的分子,因此假设这些分子有相似的形状、结合模式、溶剂效应,在静电上的差异是它们的主要差别,这使得EC特别适合用于快速评估。结果表明,EC score与实验值具有显著的线性关系,Peason R2=0.45。EC是基于Cresset XED力场,不能评估色散等相互作用成分,所以进一步用SQM进行更全面的相互作用能计算。结果发现SQM ΔE的Peason R2=0.81,RMSE=0.78,活性与非活性得到完美的区分,性能可与重计算量的FEP方法相当。这个计算过程非常快速,根据结果可将打分高的化合物送入Make与Test周期加速运转。
Flare Docking打分函数LF VSscore在精心准备蛋白结构的前提下,也获得相当好的结果,Peason R2=0.87;但是预测误差较大,RMSE=2.36 kcal/mol,这使得LF VSscore仅适合于药物发现漏斗的早期。在ACK1数据集中,LF VSscore完美地区分了活性与非活性化合物,为后续步骤提供了足够好的富集性能,甚至可以直接进入Make与Test周期。
总的来说,Flare EC与LF VSscore可以快速、高效地富集活性化合物,减少非活性化合物进入后续重型计算,加速DMTA周转而加快药物药发现。
方法
分子叠合、分子对接与EC打分
蛋白结构准备、分子叠合、分子对接、EC打分、SQM(xTB)几何优化采用Flare软件8。
统计与线性回归拟合
统计与线性回归拟合采用python软件包scipy9的stats模块来计算。
SQM相互作用能计算
SQM ΔE采用xTB V6.7.0软件包在GFN2-xTB理论水平计算6,10。
结论
本文深入分析了Flare两种轻量级LF VSscore与EC score打分方法对ACK1激酶抑制剂铰链结合片段优先级排序的性能,旨在为骨架跃迁的不同母核发现漏斗提供高效、准确的方法。Flare静电互补性打分(EC score)作为一种快速的筛选工具,揭示了抑制剂铰链结合片段与ACK1激酶结合位点间的静电相互作用对活性的影响,其与实验数据的显著线性关系初步验证了静电差异作为活性差异的主要原因。相互作用成分更完整的SQM ΔEinter打分进一步提高了对不同母核的排序能力,表现出与实验值的高相关性。Flare的LF VSscore虽然在预测精度上存在一定的误差,但其在活性与非活性化合物的区分上表现出色,为药物发现的早期阶段提供了有效的富集手段。总的来说,Flare的LF VSscore与EC score提高了筛选的效率,减少了非活性化合物进入后续重型计算,加速了DMTA运转进而加快药物的发现。
附件
- ec_xtb_sqm_score: EC与xTB计算SQM ΔE
- LF_VS-score: Flare docking打分
- 4ewh_gist_analysis: GIST分析结果
文献
- Jiao, X. et al. (2012) “Synthesis and optimization of substituted furo[2,3-d]-pyrimidin-4-amines and 7H-pyrrolo[2,3-d]pyrimidin-4-amines as ACK1 inhibitors,” Bioorganic and Medicinal Chemistry Letters, 22(19), pp. 6212–6217. Available at: https://doi.org/10.1016/j.bmcl.2012.08.020.
- Granadino-Roldán, J.M. et al. (2019) “Effect of set up protocols on the accuracy of alchemical free energy calculation over a set of ACK1 inhibitors,” PLoS ONE, 14(3). Available at: https://doi.org/10.1371/journal.pone.0213217.
- 多之“毫厘”,一个水分子显著提升FEP精度|靶点“探月”计划. Hermite药物设计. 微信公众号. 2022年06月30日,18:30,北京
- Bauer, M.R. and Mackey, M.D. (2019) “Electrostatic Complementarity as a Fast and Effective Tool to Optimize Binding and Selectivity of Protein–Ligand Complexes,” Journal of Medicinal Chemistry, 62(6), pp. 3036–3050. Available at: https://doi.org/10.1021/acs.jmedchem.8b01925.
- 肖高铿.用静电分析TYK2 JH1抑制剂铰链结合片段的SAR. 墨灵格的博客. 2023-02-19. Available at: http://blog.molcalx.com.cn/2023/02/20/tyk2-inhibitor-pf-06826647.html
- Bannwarth, C., Ehlert, S. and Grimme, S. (2019) “GFN2-xTB – An Accurate and Broadly Parametrized Self-Consistent Tight-Binding Quantum Chemical Method with Multipole Electrostatics and Density-Dependent Dispersion Contributions,” Journal of Chemical Theory and Computation, 15(3). Available at: https://doi.org/10.1021/acs.jctc.8b01176.
- Pecina, A. et al. (2024) “SQM2.20: Semiempirical quantum-mechanical scoring function yields DFT-quality protein–ligand binding affinity predictions in minutes,” Nature Communications, 15(1). Available at: https://doi.org/10.1038/s41467-024-45431-8.
- https://www.cresset-group.com/software/flare
- https://scipy.org
- https://github.com/grimme-lab/xtb


