
摘要:Flare的静电分析技术包括蛋白静电相互作用势(PIP),配体场与静电互补性(EC)分析。在本文文中,以辉瑞公司的TYK2 JH1抑制剂PF-06826647的铰链结合片段的优化为例,演示如何用Flare的静电分析方法来研究、理解不同母核TKY2 JH1抑制剂的SAR,并对这些母核进行排序、优选。
肖高铿/2023-02-10
前言
辉瑞公司的Gerstenberger等人1报道了Ropsacitinib(PF-06826647, Compound 22)的发现过程。PF-06826647是一种选择性的Tyrosine kinase 2 (TYK2) 的口服抑制剂,结合到TYK2催化活性JH1结构域。当[ATP]=1mM时,PF-06826647抑制TYK2、JAK1、JAK2的IC50值分别为17nM、383nM与74nM。目前PF-06826647已经针对溃疡性结肠炎、银屑病和化脓性汗腺炎等适应症开展II/III期临床试验。
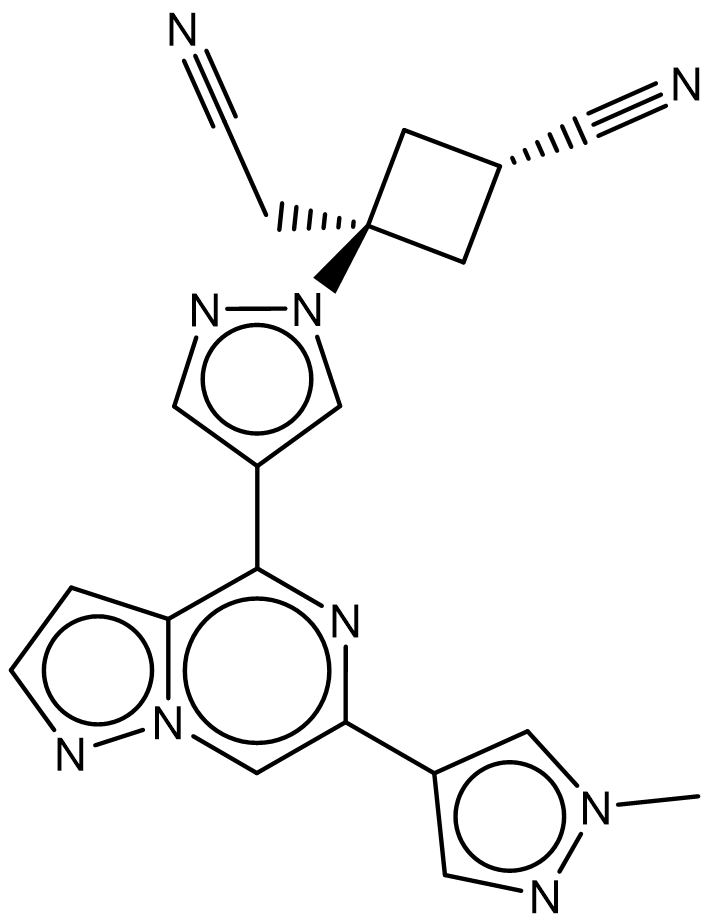
图1. 化合物22(PF-06826647)的化学结构式
在对绞链结合片段母核的优化过程中,考察了图2(上,蓝色部分)所示化合物8-11的四种不同母核。其中五元芳环1位的N作为氢键受体、六元芳环7位C-H作为氢键供体与TYK2的Val981发生氢键相互作用。
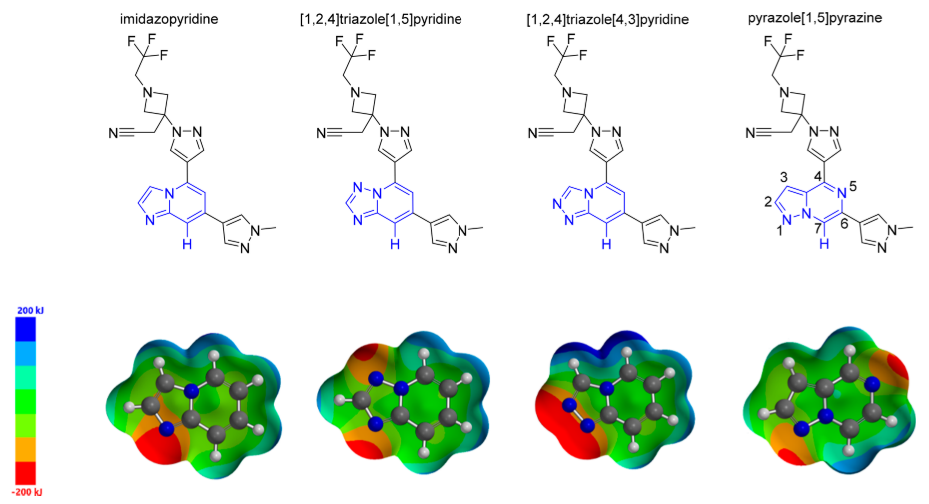
图2. 上:从左到右依次为化合物8-11的化学结构式;下:对应绞链结合片段的静电势。图片来源于Gerstenberger等人1的文章。
用量子力学(QM)方法在HF/6-31G*理论水平计算了四个母核的静电势,结果如图2(下)所示:红色表面对应负静电势区,蓝色表面对应正静电势区。可以发现,四个铰链区结合片段母核的1位N都对应一个代表负静电势的红色表面,而7位C-H对应着代表正静电势的蓝色表面。化合物的这种静电特征有利于与TYK2 Val981发生氢键相互作用。如图3所示的相互作用模式所示,1位N的负静电势与Val981的酰胺NH互补,发生经典的氢键相互作用;而7位正静电势的C-H指向Val981的羰基氧,发生C-H···O=C氢键相互作用。

图3. 铰链结合片段母核与Val981的相互作用模式,图片来源于Gerstenberger等人1的文章。
尽管化合物8-11的铰链结合片段母核的1位N与7位C-H在静电特征上相似且都与Val981的酰胺NH及羰基O静电互补发生氢键相互作用,但是它们在ATP浓度为1mM时对TYK2的酶学活性(IC50)具有显著差异,如表1所示。
表1. 化合物8-11的TYK2活性与性质
| Items | 8 | 9 | 10 | 11 |
|---|---|---|---|---|
| TYK2 IC50(nM)a | 290 | 476 | 大于10000 | 10 |
| sfLogD | 2.1 | 2.3 | 1.2 | 2.5 |
| TYK2 LipE | 4.5 | 4.0 | 小于3.8 | 5.5 |
a. 酶学测试,取两次测试的平均值,ATP的浓度为1mM。
比较四个母核的QM静电特征与活性,显而易见的,这种活性差异与母核在其它位置存在静电特征差异有关。本文的主要目的是用Flare V6.1的静电分析技术,从配体角度与蛋白角度来分析构效关系,从而对母核进行理性地设计、排序与优选。
数据的准备
化合物11与TYK2 JH1结合的蛋白质-配体共晶结构从Protein Data Bank下载(PDB:6X8G)并在Flare中精心准备。在Flare 3D视窗中检查分子结构的互变异体和质子化状态,然后将X-衍射线晶体结构PDB 6X8G的共晶配体(化合物11)提取到配体表单。在配体表单里复制化合物11,并在结合位点里对11进行原子替换得到结合构象的化合物8、9、10,对铰链结合片段的母核进行力场优化后备用。删除8-11的侧链,则得到化合物8-11结合转态的母核结构。
结果
Vina分子对接打分函数不能区分8-11的活性差异
AutoDock Vina分子对接方法是常用的虚拟筛选与结合亲和力预测方法2。Gaillard3的研究表明证明,在score_only模式下,比之绝大部分分子对接软件的打分函数,Vina的亲和力预测值与实验值具有最好的相关性。用Vina在TYK2-11共晶结构(PDB 6X8G)的JH1“干”结合位点里计算化合物8-11的亲和力,结果如表2所示。
表2. 用AutoDock Vina 1.2打分函数预测8-11对与TYK2 JH1的结合亲和力
| Items | 8 | 9 | 10 | 11 |
|---|---|---|---|---|
| gauss 1 | 127.64897 | 127.03038 | 127.26082 | 126.18600 |
| gauss 2 | 1635.64079 | 1635.32122 | 1635.86922 | 1635.60318 |
| repulsion | 2.29394 | 2.28174 | 2.21230 | 2.29136 |
| hydrophobic | 34.25257 | 34.25257 | 34.25257 | 23.20991 |
| H-Bond | 1.22853 | 1.22853 | 1.22853 | 1.22853 |
| Vina Affinity(kcal/mol) | -10.51240 | -10.50150 | -10.55757 | -10.66299 |
| pIC50 | 6.54 | 6.32 | 5 | 8 |
显而易见的,8-11具有相似的结合亲和力预测值,不能将高活性化合物11与另外3个低活性化合物区分开来;此外预测值(Affinity score)与实验值(pIC50)之间没有显著的线性关系,Pearson相关性系数R2=0.376。总的来说,Vina打分值不能对8-11的活性进行排序,不能仅依靠Vina打分值来优选母核结构。
化合物8-11与TYK2的静电互补性(EC)与活性(pIC50)呈线性关系
Bauer等人4提出的静电互补性打分(EC Score)的基本思想是:当配体和受体的静电势匹配时(即具有相同的量值和相反的符号),实现配体和受体之间的最大静电亲和力。已经在很多数据集上做过测试4,EC打分值与活性值之间具有从中等程度到非常好的相关性。用Flare静电互补性(EC)对8-11进行打分,可以观察到pIC50与EC_rho具有极其显著的相关性,Pearsion线性相关性系数R2=0.91,如图4所示。
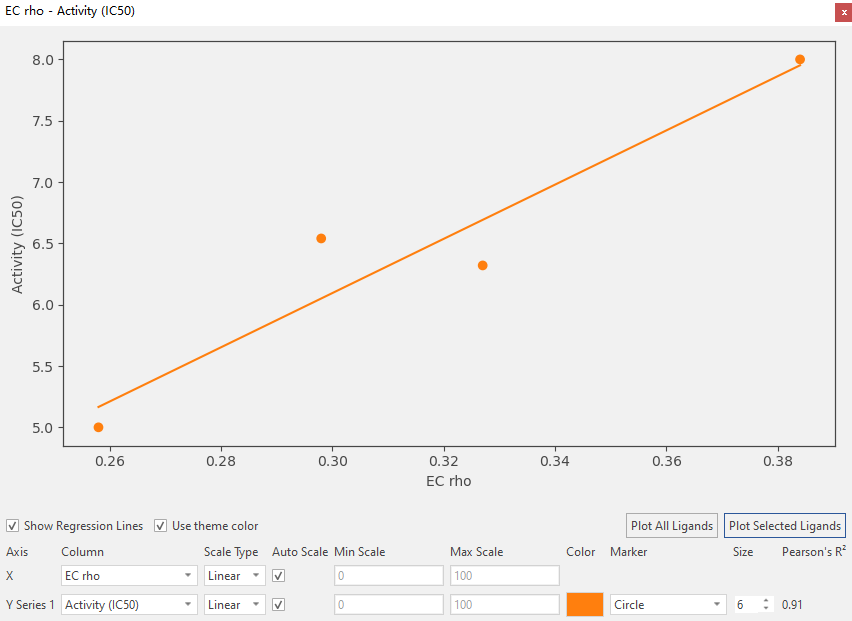
图4. 化合物8-11的pIC50与EC之间具有显著的线性相关性
绘制8-11的分子表面并用EC值着色,结果如图5所示,可以看到在铰链结合片段母核5位处,化合物11呈现互补的绿色,而其它化合物8-10呈现红色。母核5位的更好静电互补决定了化合物11的活性要高于其它化合物。
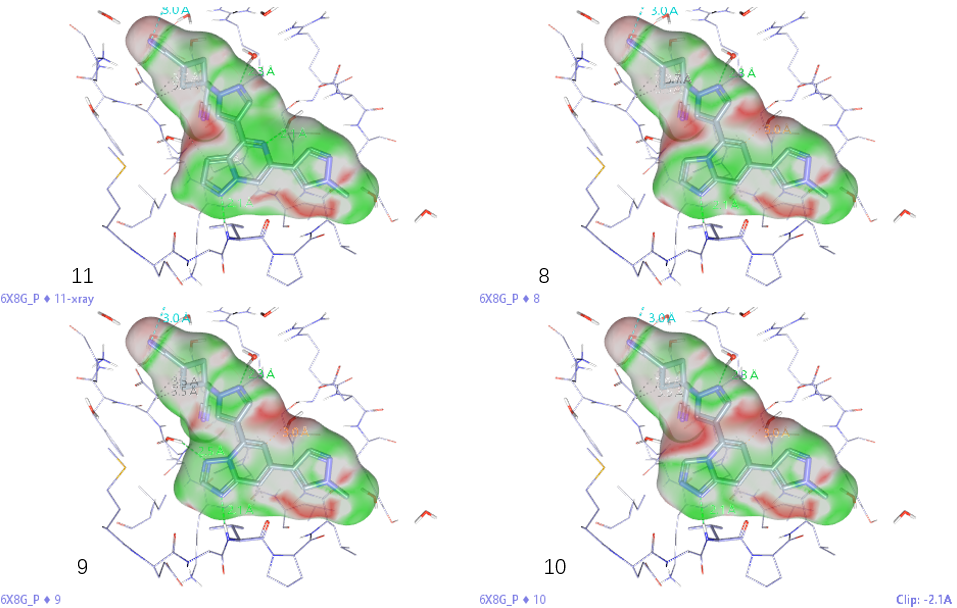
图5. 化合物8-11的分子表面,用静电互补性(EC)着色:绿色——EC互补;红色——EC不互补。
母核与TYK2的静电互补性与活性成线性关系
相似的,我们可以观察到化合物8-11的母核静电互补性与pIC50也存在线性关系,如图6所示,Pearsion线性相关性系数R2=0.898。
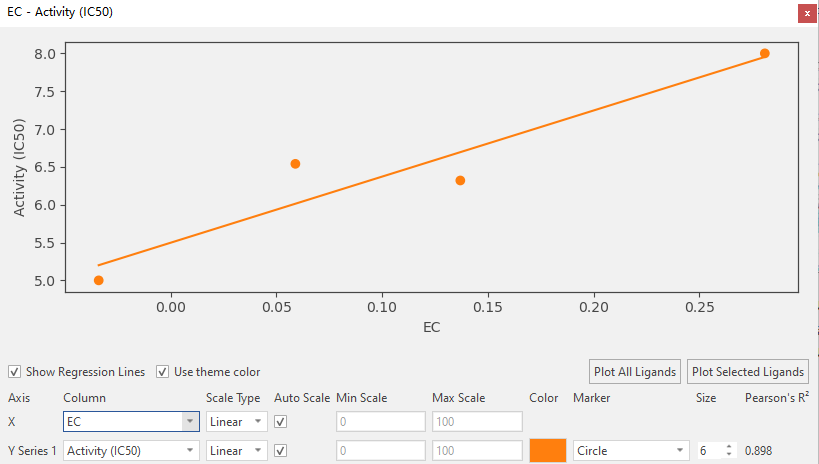
图6. 化合物8-11的pIC50与母核EC之间具有显著的线性相关性。
绘制8-11的铰链结合片段母核分子表面并用EC值着色,结果如图7所示,在母核5位处,化合物11呈现互补的绿色,而其它化合物8-10呈现红色。再一次,我们看到了母核5位的更好静电互补决定了化合物11的活性要高于其它化合物。
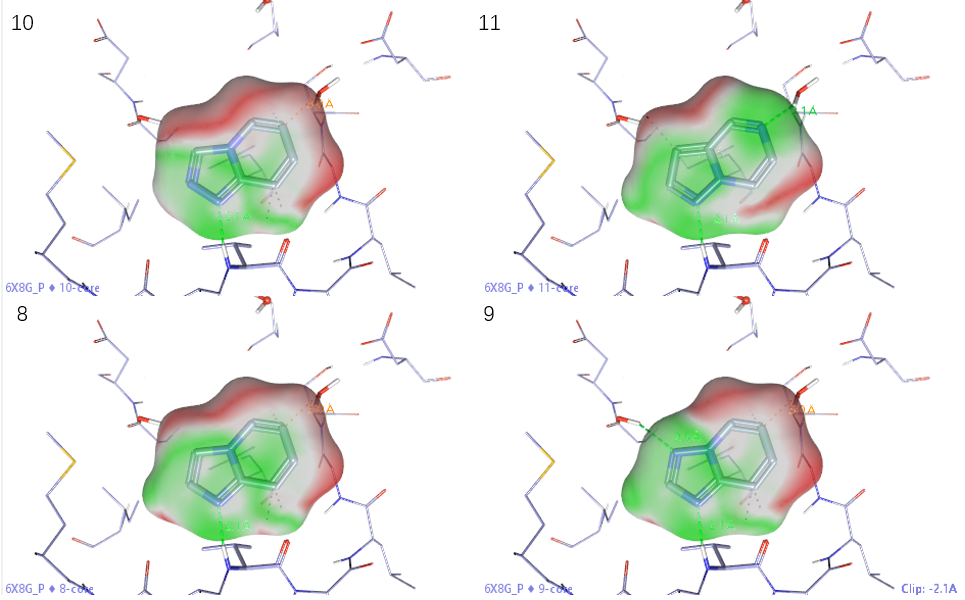
图7. 化合物8-11的铰链结合片段母核分子表面,用静电互补性(EC)着色:绿色——EC互补;红色——EC不互补。
比较蛋白相互作用势(PIP)与配体场
PIP提供了蛋白结合位点详细的静电作用图,因此可以为理解配体结合、SAR和靶向蛋白的新分子设计提供非常有用的见解。可视化比较PDB 6X8G活性位点的PIP(图8左)与化合物11(pIC50=8,四个化合物中活性最强的)的配体场,可以看到化合物11母核5位右侧的负静电场刚好与TYK2活性位点的正PIPs重合(图8右)。同时,从图8右可以发现,配体11的1位下方的负配体场与TYK2活性位点Val981酰胺NH对应的正PIP重合,配体7位C-H下方的正配体场覆盖了TYK2活性位点Val981的羰基氧。化合物11与TYK2的静电互补图也证实了配体场和蛋白静电之间的良好互补性(图5,左上角)。
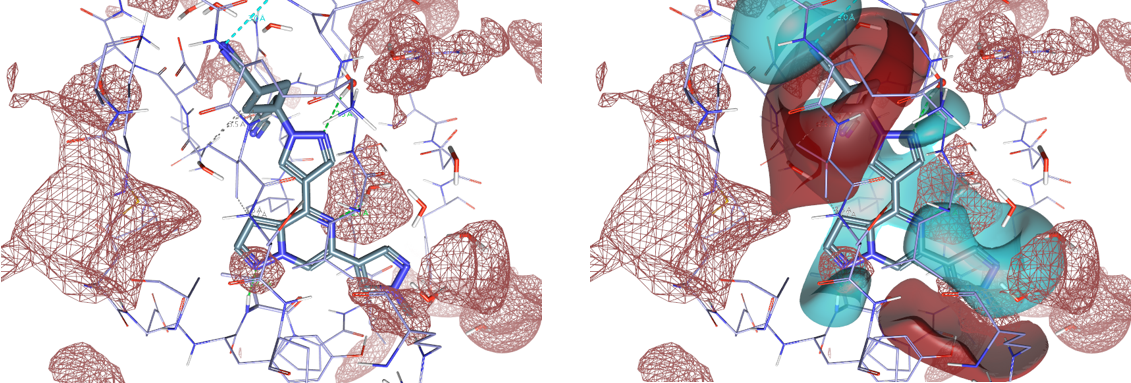
图8. 左:蛋白TYK2(PDB 6X8G)JH1结合位点的正PIP;右:蛋白TYK2的正PIP与化合物11的配体场比较。
相比之下,化合物9-10的铰链区结合片段母核5位处红色正的配体场(图9,实心红色区块)与右侧蛋白TYK2的红色正PIP(图9,网格红色区块)重合,这意味着化合物9-10在该处与蛋白TYK2发生静电冲突。从图5的静电互补图也可发现,在化合物8、9、10的5-位处为红色,这是静电不互补的表现。
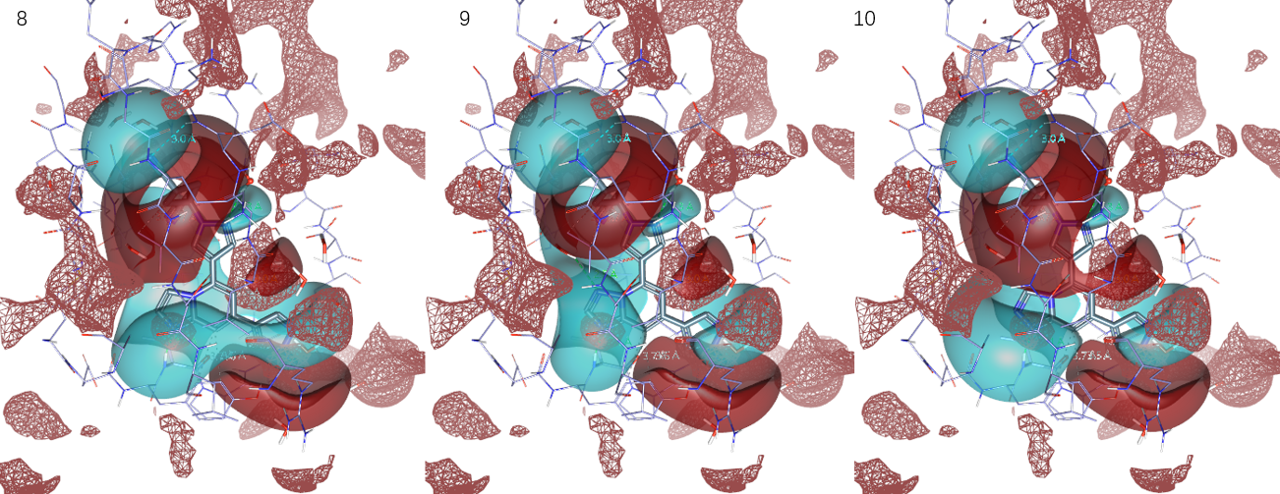
图9. 蛋白TYK2(PDB 6X8G)JH1结合位点的正PIP(网格状红色区块)与化合物8、9、10的配体场比较(红色实心区块:正静电场;蓝色实心区块:负静电场)。
总的来说,化合物8-11的配体场与TYK2结合位点PIP的互补性与前面静电互补性基本一致,也与8-11的活性高低也相一致。
用配体场分析C-H···O=C氢键相互作用
尽管Flare内置了QM方法PSI4可以用于计算配体的静电场以重现Gerstenberger等人1用DFT方法分析C-H氢键供体的结果,但计算速度很慢;基于XED力场的配体场可以重现DFT方法计算的静电势、快速地完成C-H氢键的分析。氢键供体和受体在官能团周围的分布是相互作用势的良好代表,并且计算获得的场点模式与该信息一致。
在Flare中,可以用两种方式对C-H氢键供体进行分析:定性可视化或定量分析。以活性最强的化合物11为例,如图10所示,可视化11的配体场并与蛋白进行比较(为方便起见,仅呈现Val981相邻的4个残基),可以发现:1位N的蓝色负配体场覆盖了Val981的酰胺NH;而7位C-H的红色正配体场覆盖住了Val981的羰基氧原子。化合物配体场特征与QM计算静电特征一致,可以正确理解11与Val981的相互作用。
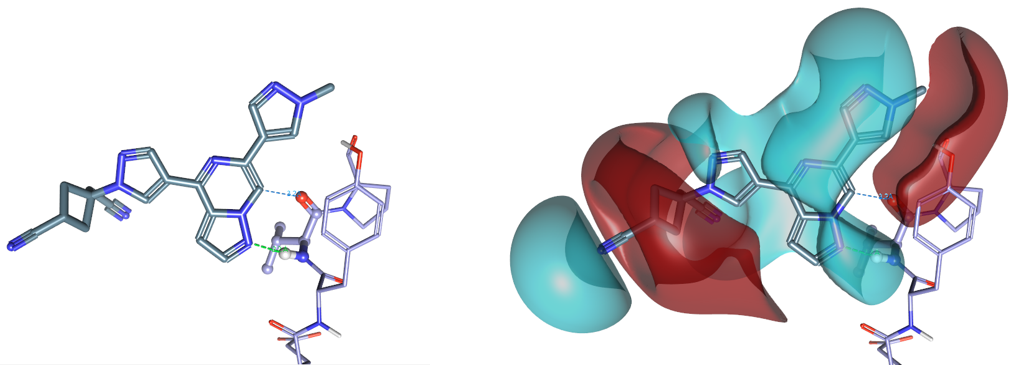
图10. 左:化合物11与Val981(球棍模型)相互作用图,右:化合物11的配体场(蓝色为负配体场,红色为正配体场)与Val981的比较
然而,在实践中我们可能需要对很多化合物(比如分子对接后产生的大量化合物pose)进行分析以判断一个化合物是否与Val981的羰基氧发生氢键相互作用,无论是通过经典氢键供体,还是通过非经典的C-H氢键供体。这个问题可以转化为:一个化合物是否在Val981羰基氧的坐标上具有正的配体场。因此,我们仅需要计算化合物在Val981羰基氧坐标上的配体场:如果场值为正,则可能存在氢键相互作用;如果场值为负,则可能静电冲突,不发生氢键相互作用。Flare提供Python API可以实现批量对空间特定坐标点的配体场计算,具体的操作过程请参考之前的分子对接后处理博客文章5。结果如表3与图11所示,化合物8-11在Val981羰基氧处的配体场值全部为正,说明化合物8-11均可能与Val981发生C-H氢键相互作用。活性最强的化合物11在Val981羰基氧处的配体场值最强,这与QM计算结果一致:化合物11在对应C-H正静电区的蓝色最为浓郁,相比之下8、9与10的蓝色非常淡。
表3. 化合物8-11在Val981羰基氧原子、酰胺NH氢原子以及Glu979羰基氧原子处的配体场值
| Items | 8 | 9 | 10 | 11 |
|---|---|---|---|---|
| Val981-C=O Field Value | 0.6311 | 1.7136 | 1.0156 | 3.1610 |
| Val981-NH Field Value | -12.1416 | -11.2555 | -12.8432 | -7.02595 |
| Glu979-C=O Field Value | -2.4217 | -1.5101 | -4.6231 | -0.7160 |
相似的,我们还可以计算与化合物8-11在TYK2 Val981酰胺NH供体氢(见图11)位置上的配体场值。我们可以预期Val981-NH氢原子上的配体场值应该为负,因为对应着化合物母核1位氮原子的孤对电子。结果如表3与图11所示,8-11在该处的配体场确实为负。还可以注意到,活性最强的化合物11在该点的场值并非最低,这与图2所示QM计算结果一致:化合物11的1位氮原子对应的红色区块面积似乎更小、颜色也没有那么浓郁。
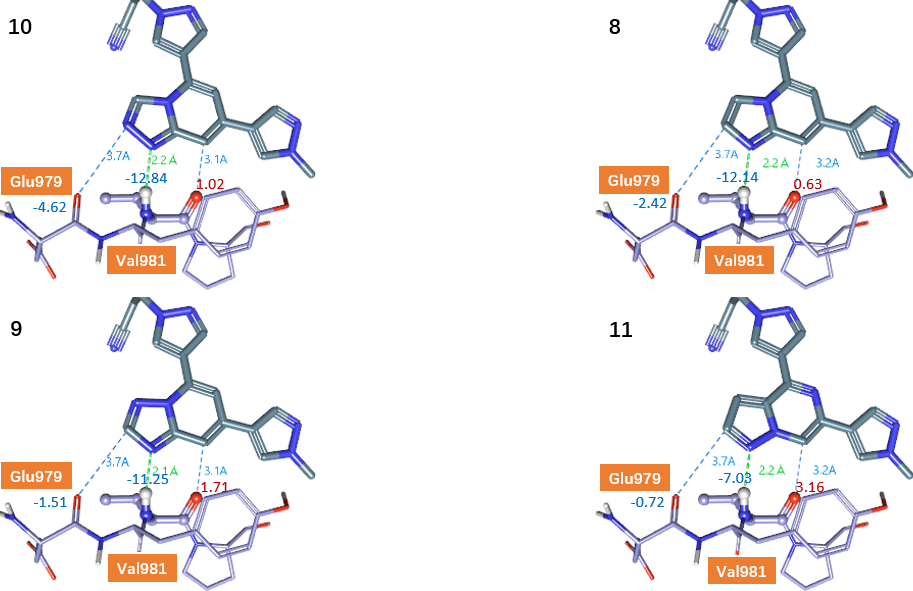
图11. 化合物8-11在Gul979羰基氧原子、Val981酰胺羰基氧原子与NH氢原子上的配体场值。蓝色数值:负配体场;红色数值:正配体场。
根据图8、9的配体场可以发现,Glu979的羰基氧靠近配体的蓝色负配体场,这让人疑心配体与Val979的羰基氧是否在静电上有冲突。因此有必要计算Glu979碳基氧处的配体场,结果如表3与图11所示:化合物8-11在该处的配体场值均为负值,这说明在该处蛋白与配体之间可能存在静电冲突。化合物8-11的活性pIC50与该点的配体场值具有极强的线性相关性,Pearson相关性系数R2=0.843。其中活性最强的化合物11在该点的配体场值(-0.72)最低,也就是静电冲突最低;活性最低的化合物10在该点的配体场值(-4.62)最大,也就是静电冲突最大。这进一步说明了场分析的优势:配体场与蛋白相互作用场(PIP)分析可以揭示经典相互作用不能发现的重要分子间相互作用。作为gatekeeper残基Glu979的C=O从经典分子间相互作用模式看似没有参与相互作用,但是从配体场角度看实则参与,甚至决定了活性强度。
总的来说,Flare可以方便地将经典的氢键相互作用与非经典的C-H···O=C氢键相互作用问题转化为统一的配体场问题进行可视化分析或数值定量分析。
用Flare QM分析Glu979-C-H···O=C氢键相互作用
Flare QM是一个基于PSI4实现的配体量子力学 (Quantum Mechanics,QM) 计算模块。可以用来对单个配体、构象系综、配体结合构象进行几何优化和单点能量计算,还可以计算和显示配体的电子密度、前线分子轨道、分子静电势,对感兴趣配体的特定旋转键进行基于QM的两面角扫描分析(QM Torsion profile)。如果你担心QM计算速度太慢,还可以通过Cresset Engine Broker™连接到本地集群或云计算设施来加速QM计算、优化计算时间。这里以化合物10、11的母核为例,用Flare QM在B3LYP/6-31+G(d)理论水平进行重现Gerstenberger等人1的QM计算。
总的计算过程如下:
- 几何优化:QM几何优化,然后叠合到化合物10、11上
- 电子密度:单点能计算获得电子密度图,并导出为10_den.dx,11_den.dx
- ESP计算:导出为10_esp.dx,11_esp.dx
- MESP计算:将diensity = 0.0004的点定义为分子表面,并给出该点的静电势(EPS)
MESP是非常重要的分子设计参数,可以用pyflare脚本给出为各种格式,并用于后续统计分析。以化合物10母核为例,这里我给出csv格式:
1 2 3 | flare_qm_mesp.py --den 10_den.dx \ --esp 10_esp.dx \ --output 10_mesp.csv |
MESP可以通过Flare的场点来可视化,比如将较强的点列出,追加到SDF:
1 2 3 | flare_qm_field_point_to_sdf.py \ --isdf 10.sdf \ --osdf 10_mesp.sdf 10_mesp.csv |
上面的命令将计算的QM场点追加到SDF文件(10.sdf)里,并另存为带有QM场点的SDF文件(10_mesp.sdf)。将带有QM场点的sdf文件读入Flare,然后通过Flare | home | Field来激活可视化。
现在以10、11的母核为例,在Flare 3D Pose | QM 对化合物在B3LYP/6-31+G(d)理论水平进行单点能计算与静电势分析,并用pyflare的将分子表面上静电势绝对值相对高的地方提取出来,并与铰链区残基一起观察,如图12所示。
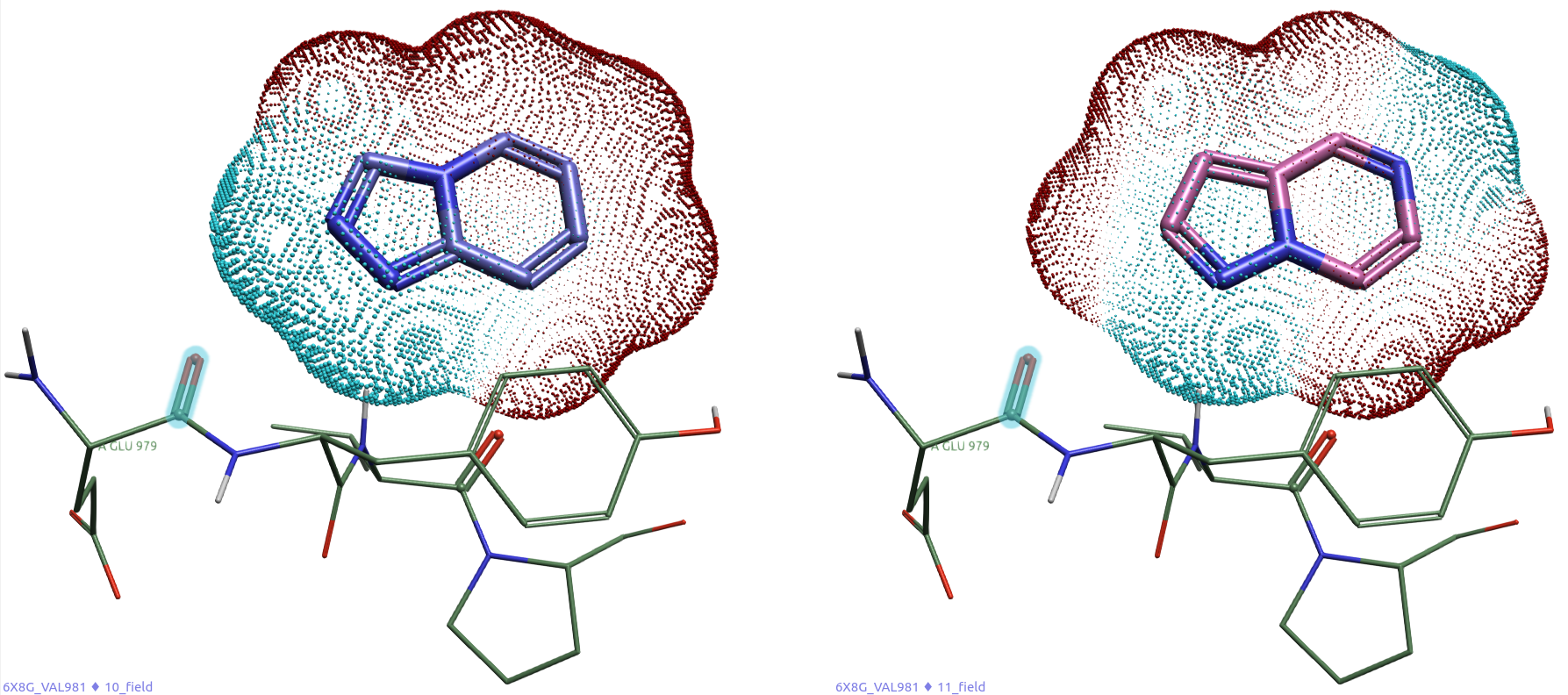
图12. 化合物10(左)、11(右)母核的分子表面静电势(MESP)。蓝色:负ESP;红色:正ESP;大小:点越大则EPS绝对值越大;高亮原子为Glu979-C=O。
可以看到,铰链区Glu979-C=O附近配体表面静电存在显著的不同,化合物10显然存在静电冲突,而化合物11则更加互补。
然而,我们对碳基氧原子能“摸”到的配体表面更感兴趣,仅显示Glu979-C=O半径范围内的配体表面,可以用来比较在Glu979-C=O碳基氧原子“眼里”10与11的差异:
1 2 3 4 5 6 | flare_qm_add_near_point_to_SDF.py --atom O \ --xyz 12.646 -11.884 11.205 \ --den 10_den.dx \ --esp 10_esp.dx \ --isdf 10.sdf \ --osdf 10_glu979_CO.sdf |
这里我用bondi原子半径来计算接触的距离(d),半径参考了Manjeera(2019)等人的文章并会汇编为字典,通过atom选项告诉脚本查寻原子半径,并计算距离(d):
$$
d = r_{atom} + 0.5
$$
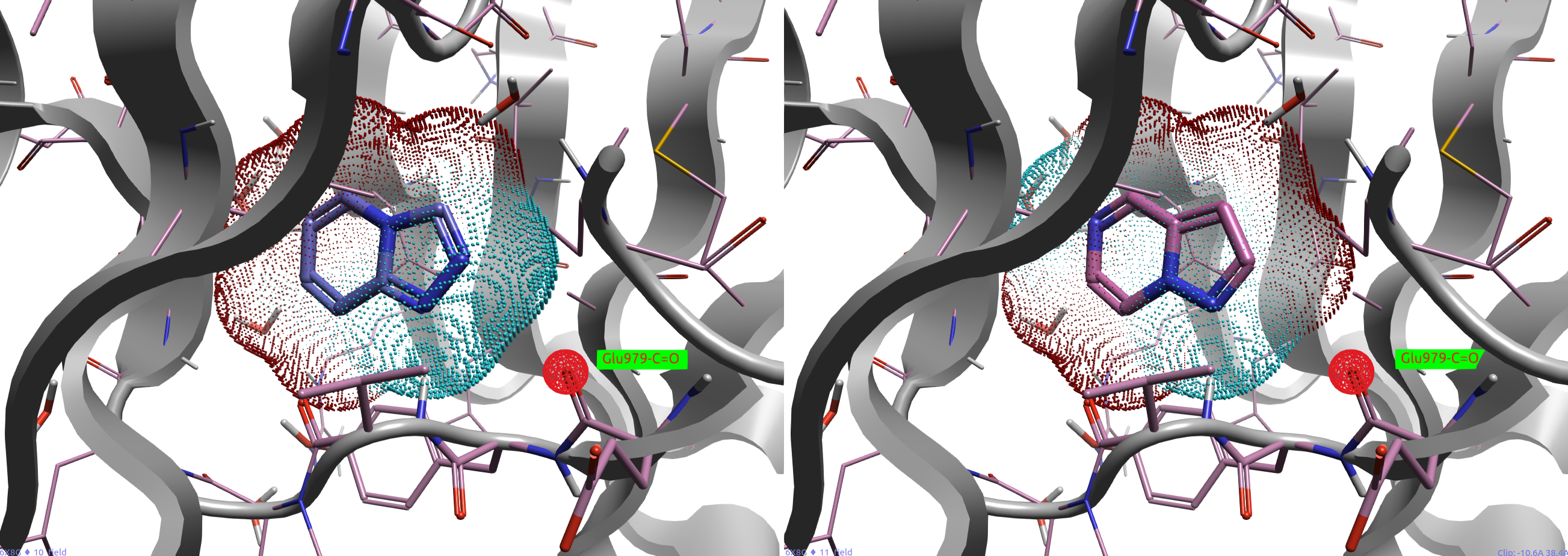
其中xyz选项是O原子的坐标。将配体表面距离Glu979-C=O在距离d范围内的QM场点追加到SDF件,读入Flare进行可视化分析,结果如图13所示。
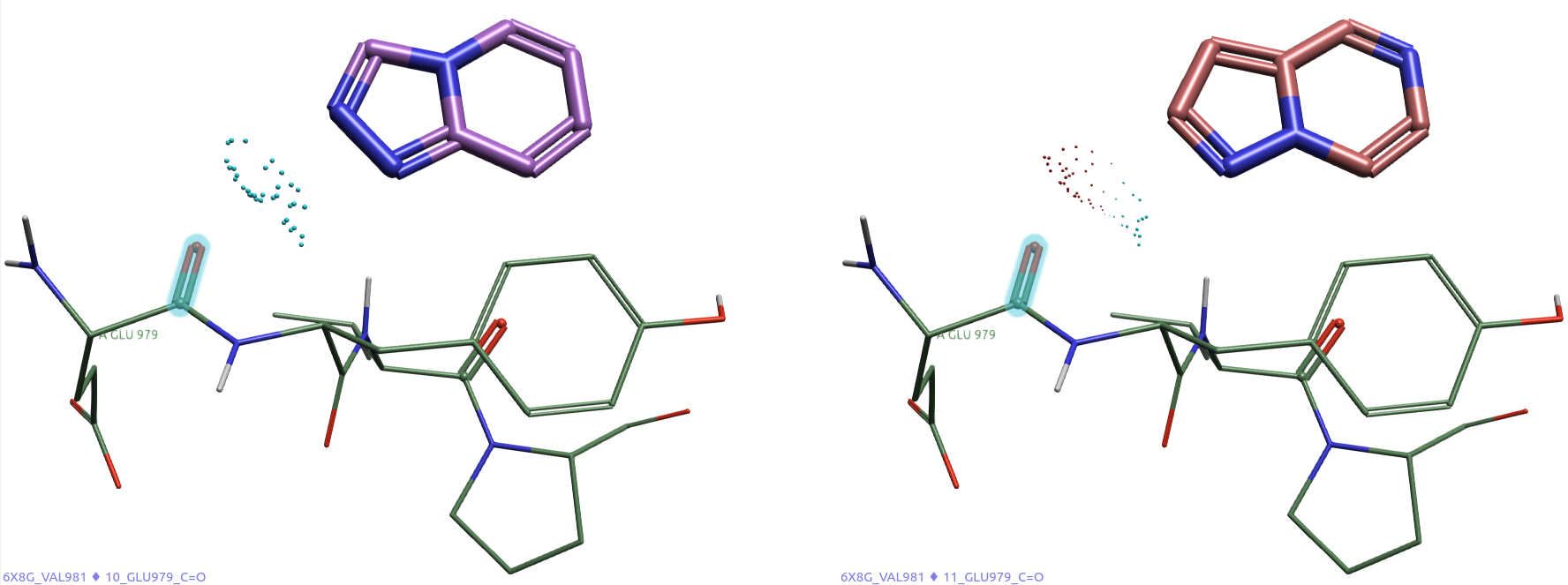
图13. Glu979-C=O能够“摸”到化合物10(左)、11(右)母核的分子表面静电势(MESP)。蓝色:负ESP;红色:正ESP;大小:点越大则EPS绝对值越大;高亮原子为Glu979-C=O。
图13是以蛋白的视角观察与之相互作用配体的特征,体现了基于结构的设计思路:蛋白摸到的配体形状与静电的样子。从图13可以看到,Glu979-C=O仅能触摸到化合物10蓝色的负静电区,也就是静电冲突;而化合物11触摸到化合物红色正静电与蓝色负静电区,以红色为主,表现出静电互补。此外,10的蓝色QM场点比11的红色或蓝色场点颗粒要更大,这也说明蛋白羰基氧与10发生的静电冲突在值上更大。所有这些都说明了化合物11比10在静电上与蛋白更加互补,这与化合物10对TYK2几乎没有活性(\(IC_{50} > 10000\ nM\))而11对TYK2表现出强的抑制活性(\(IC_{50} = 10\ nM\))的实验结果是一致的。
在药物化学实践中,配体表面的QM静电势还有其它非常重要的研究价值。比如你可以用上面的脚本找出分子表面上某个原子贡献的MESP极值,该MESP极值可用来计算pKa、探索反应选择性等等,这里就不再详细叙述。
结论
Flare的静电分析技术包括蛋白静电相互作用势(PIP),配体场与静电互补性分析。在本文中,成功地用这些静电分析方法来研究、理解不同母核TKY2 JH1抑制剂的SAR,并对这些母核进行可视化分析、排序与优选。
Flare QM提供了基于QM的配体静电分析技术,可以用更高级别的理论来研究配体的SAR,并进行可视化分析、排序与优选。此外,在药物化学实践中,Flare QM计算的MESP还可用来计算原子的pKa、探索反应选择性等等。
相关数据
- AutoDock Vina分子对接输入文件与结果:docking.zip,提取码:bgmb
- 静电分析Flare项目文件:TYK2-EC-6X8G.flr,提取码:gnyx
文献
- Gerstenberger, B. S.; Ambler, C.; Arnold, E. P.; Banker, M.; Brown, M. F.; Clark, J. D.; Dermenci, A.; Dowty, M. E.; Fensome, A.; Fish, S.; et al. Discovery of Tyrosine Kinase 2 (TYK2) Inhibitor (PF-06826647) for the Treatment of Autoimmune Diseases. J. Med. Chem. 2020, 63 (22), 13561–13577. https://doi.org/10.1021/acs.jmedchem.0c00948.
- Trott, O.; Olson, A. J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2009, 31 (2), 455–461. https://doi.org/10.1002/jcc.21334.
- Gaillard, T. Evaluation of AutoDock and AutoDock Vina on the CASF-2013 Benchmark. J. Chem. Inf. Model. 2018, 58 (8), 1697–1706. https://doi.org/10.1021/acs.jcim.8b00312.
- Bauer, M. R.; Mackey, M. D. Electrostatic Complementarity as a Fast and Effective Tool to Optimize Binding and Selectivity of Protein-Ligand Complexes. J. Med. Chem. 2019, acs.jmedchem.8b01925. https://doi.org/10.1021/acs.jmedchem.8b01925.
- 肖高铿. 分子对接后处理——与指定残基发生特定相互作用配体的过滤. 墨灵格的博客. http://blog.molcalx.com.cn/2021/11/02/docking-post-filtering.html
在药物设计过程中Flare全程陪你
Flare交付先进的科学方法、分析工具和直观、易用的增强功能,洞察您的配体-蛋白质复合物结构。
想要尝试Flare信息丰富、用户友好界面,发现它如何帮助您自信地推动潜在先导化合物优化?请现在就联系我们安排试用,快速访问Flare的广泛功能。我们的专业团队随时准备通过安装和设置为您提供支持,而我们全面的教程库——涵盖从常见工作流程到高级方法和功能的所有内容将帮助您开始使用。我们在这里帮助您更快地实现目标,让您设计出重要的分子。
- 电邮:info@molcalx.com
- 电话:020-38261356


