
分子对接后处理——与指定残基发生特定相互作用配体的过滤
摘要:分子对接后处理可以提高分子对接虚拟筛选的性能。本文分享了如何用Flare的Python API来实现可视化分析的自动化处理,以帮助大家方便地进行分子对接或其它虚拟筛选后处理,过滤出与特定残基发生相互作用的化合物。更具体地讲,本文以LigGrep的数据集为例,来说明如何用Flare的Python API来对与残基GLY863发生氢键相互作用、与残基TYR896发生pi-pi堆积相互作用的配体打标签,最后通过对标签的过滤,实现从虚拟筛选的结果里过滤出与特定残基发生特定类型相互作用的配体。还演示了如何调用RDKit中的MMFF94力场对对接结果进行重原子约束的力场优化以改善氢的几何。
肖高铿/2021-11-02
1. 前言
分子对接是基于结构的药物发现的主要方法之一。分子对接方法通常用打分值来量化化合物与蛋白之间的相互作用强度或对结合模式进行排序。为了实现分子对接的效率,会通过一个简单的打分函数来量化配体-蛋白之间的相互作用。但是这种简化的蛋白-配体相互作用模型是该方法的一个很大的限制,表现在对pose排名的不准确,并且在预测绝对或相对结合自由能方面表现不佳。此外,配体结合模式的正确采样可能受限于诱导契合效应以及蛋白质的不同构象状态,这些因素导致的不精确度超过打分函数自身所带来的不精确度。
Fishcer等人[1]对文献的调研以及对93位来自学术与制药工业的专家问卷调查表明,人们基本达成共识:成功的分子对接虚拟筛选需要一定的人工后处理。其中最值得注意的是,打分函数产生的对接分数最不受重视,特别是对制药行业的专家来说。看来,已知的对接算法打分和排序的不可靠性使其在专家中不大受欢迎。互补性检查,氢键相互作用以及与特定残基相互作用在后处理中最受重视。
本文收集了一些分子对接后处理提高虚拟筛选性能的文献[2-9]供大家参考。为了让后处理更简单些,人们开始开发通用的后处理过滤程序,比如Ha等人[10]开发的LigGrep以及Gushchina等人[11]开发的VsFilt。
本文的主要目的是为大家提供一个可视化分析自动化的实现方法以帮助大家方便地进行分子对接或其它虚拟筛选的后处理,以便过滤出满足特定相互作用条件的化合物。
本文以Ha等人[10]的LigGrep所带的算例来演示如何用Flare的Python API进行对接后处理。更具体地讲,如图1所示,本文将演示如何(1)过滤出GLY863发生氢键相互作用的配体;(2)过滤出与TYR896发生pi-pi堆积的相互作用的配体。我相信,在这个算例启发下很容易举一反三地实现其它相互作用的过滤后处理。
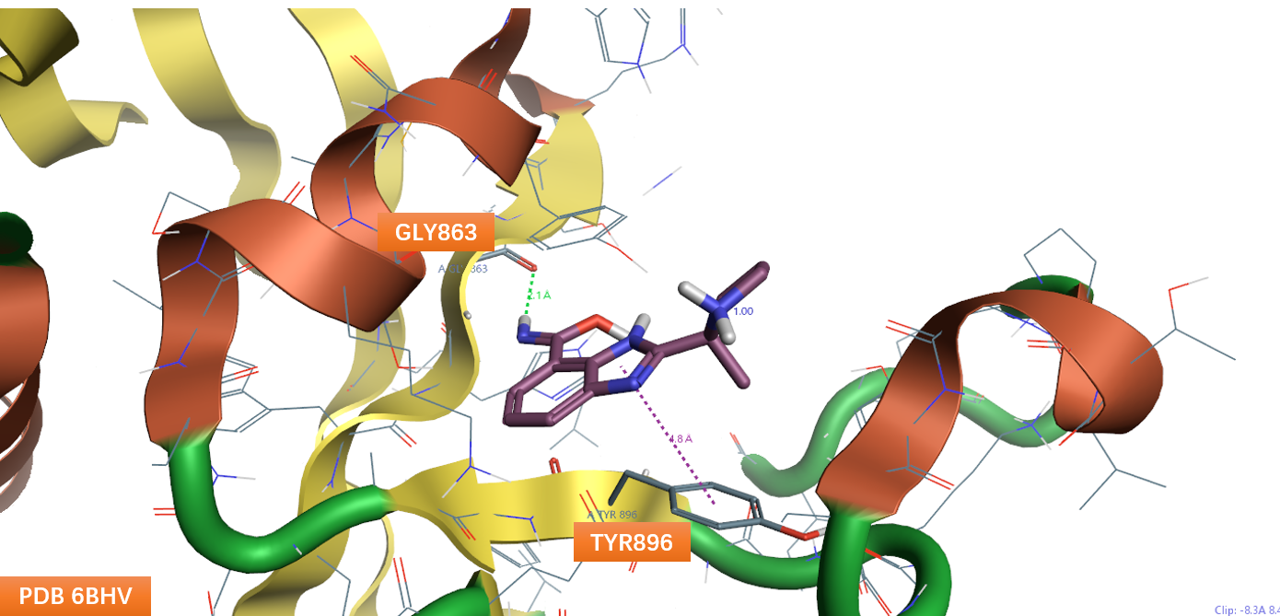
图1. 以LigGrep的算例来说明:如何过滤出与GLY863发生氢键相互作用的配体,与TYR896发生pi-pi堆积相互作用的配体。
再次提醒:这只是个演示,在实际研究中应该根据需要选择过滤条件。
2. 数据与方法
2.1 数据集
在Ha等人[10]开发的LigGrep软件包中提供了一个算例,研究结果表明,使用LigGrep进行过滤之后大大提高了Vina虚拟筛选的性能。该算例提供PDBQT与SDF两种格式的对接结果,在本文中被将sdf文件读入到Flare后再进行分析。
数据集的准备:将LigGrep数据集[11]的sdf文件合并,读入到Flare里,质子化状态保留了输入文件的样子,并对分子按出现出现的顺序重新命名:如果是ligand分子则以ligand开头并加上下划线与序号;如果是decoy分子则以decoy开头并加上下划线与序号。LigGrep数据集的蛋白结构没有链的信息,4个链被当成一个链混合在一起,此外,其坐标比之PDB上的坐标也发生了移动。为了方便后续的比较研究,我将蛋白的坐标根据Cα进行叠合、移动到PDB 6BHV的A链坐标上,AutoDock Vina的对接pose也跟着蛋白Cα一起移动到PDB 6BHV的A链坐标上。没有进行其它任何处理,除了坐标迁移之外,所有的东西都与原文一致。此外,为了方便按Vina score排序,还将REMARK列改名为Vina_score,将Vina打分值提取出来替换了原先的文本内容。
需要注意的是LigGrep的数据集是Vina的docking结果,在vina的docking过程中,并没有利用极性氢的方向[12]。在生成的pose中,其极性氢是随机加上去的,方向与两面角没有任何实际意义。以Decoy_46为例(如图2所示),羟基与氨基上氢发生了空间立体冲突(注意:对VINA来说,这并非错误,也不会影响它的性能),右边的平面六员环与其中一个SP3杂化碳原子不匹配,估计该六员环应该是个吡啶。
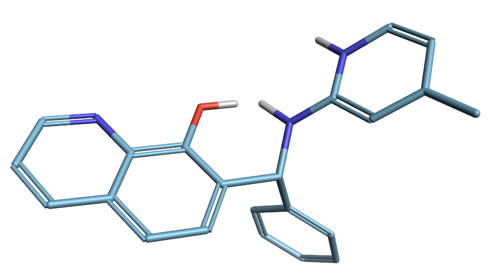
图2. decoy_46是一个不合理极性氢取向与不合理键级的例子
因此,从LigGrep读入的算例可能有很多异常的地方,对之视而不见就可以了。虽然读入的氢会影响Flare对氢键的计算,但是出于演示的目的,本文忽略了这种结构上的不合理,当成是正确的用来演示如何进行后处理。正因为如此,用Flare Viewer分析Vina的氢键相互作用可能与LigGrep就会不同。
千万注意:在正式的研究中,使用Flare Viewer再打分或分析之前应该对读入的pose在蛋白背景下重新加氢、优化。关于氢的处理,第4小节演示了如何用MMFF来进行优化。
点击下载演示用的LigGrep数据集:liggrep.flr。
2.2 用Flare Viewer来人工检查蛋白-配体相互作用
Flare Viewer是一款免费的可视化软件,Flare Viewer的Contact支持的可视化相互作用分析包括:
- 氢键
- 立体碰撞
- 阳离子-pi
- 硫-孤对电子
- 卤键
- 芳香-芳香堆积
- 盐桥/金属螯合
2.3 用Flare Python API进行自动化分析
Flare的Python API提供了cresset.flare.contacts来分析各种相互作用,以帮你从分子对接结果中过滤出满足条件的分子或pose。这些相互作用包括:
- aromatic_aromatic
- cation_pi
- h_bonds
- halogen_bonds
- steric_clashes
- sulfur_lone_pair
- salt_bridge
使用方法很简单,比如返回一个分子列表里所有分子内与分子间的芳香相互作用:
1 | cresset.flare.contacts.aromatic_aromatic(molecule list) |
再比如,返回protein与ligand的分子内与分子间所有氢键相互作用:
1 | h_bond = cresset.flare.contacts.h_bonds(protein1,ligand1) |
这些相互作用还有三个属性:points(相互作用由哪些原子组成),value(对于距离、角度与两面角的测量,还有对应的值),strength(强度,比如氢键强度有weak,average与strong三个级别强度的区分)。我们可以将属性值标注到每个对接结果里,以便进行进一步的分析。
实现上面要求的代码很简单:
3. 结果
复制上面代码到Flare python界面里,或者在命令行执行该代码,则完成:
- 在与GLY863发生氢键相互作用的配体上添加标签:GLY863_HB
- 在与TYR896发生pi-pi堆积的相互作用的配体上添加标签:TYR896_Pi_Stack
观察Flare分子表单的Tags列,发现有的分子添加了GLY863_HB标签,有的添加了TYR896_Pi_Stack标签,有的两者都添加了,如下图3所示。
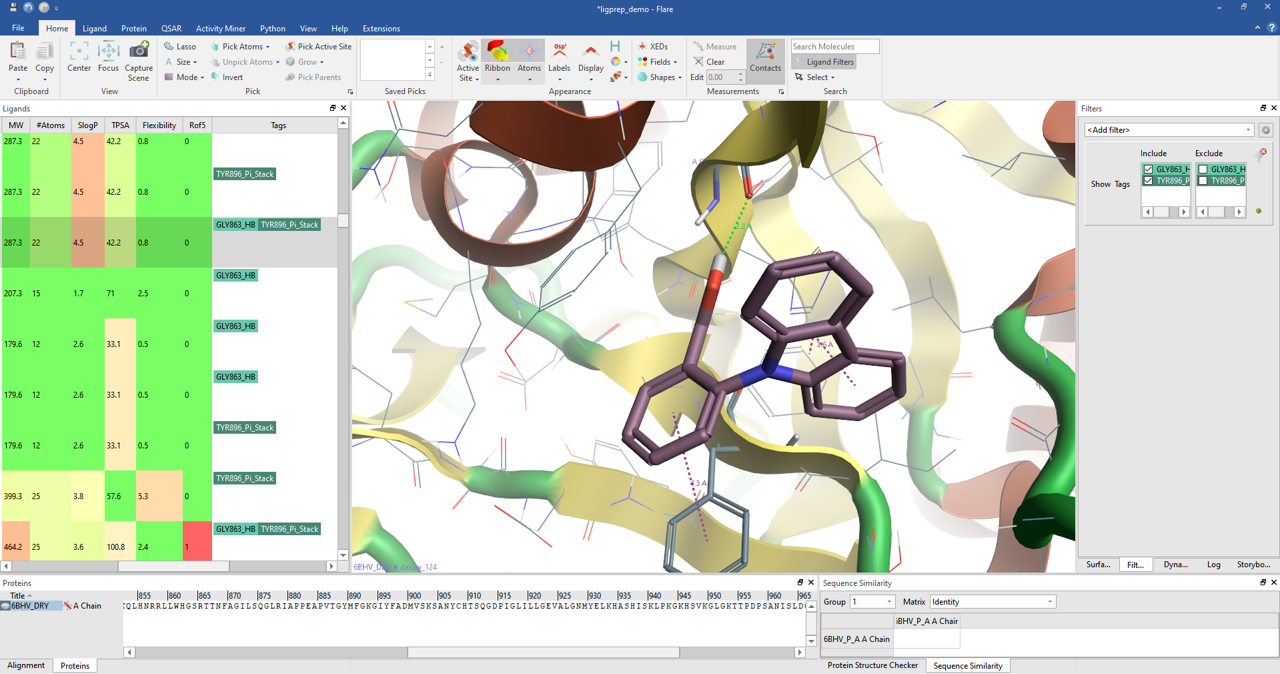
图3. 执行代码,配体表单的变化以及Tags标签的过滤
现在,就可以在Filter小部件里对配体表单进行过滤,如图2右上角所示。这样,我们就很方便完成对docking结果的后处理:过滤出与特定残基发生特定相互作用的配体。
以化合物Ligand_22为例,如图4所示,在tags列(最右边)同时包含了GLY863_HB与TYR896_Pi_Stack标签,这说明Flare Python认为化合物ligand_22与GLY863发生了氢键相互作用,同时又与TRY896发现pi-pi相互作用。这一点可以从Flare Viewer的3D视窗中可以得到验证:配体的亚胺基的氢-C=N-H作为供体与GLY863的羰基发生氢键相互作用(绿色虚线表示);配体的吡嗪环与蛋白TRY986发生pi-pi相互作用(紫色虚线表示)。这说明,Python API的分析结果与视窗呈现的相互作用一致,脚本可以代替人工分析过滤出与指定残基发生特定作用模式的化合物。
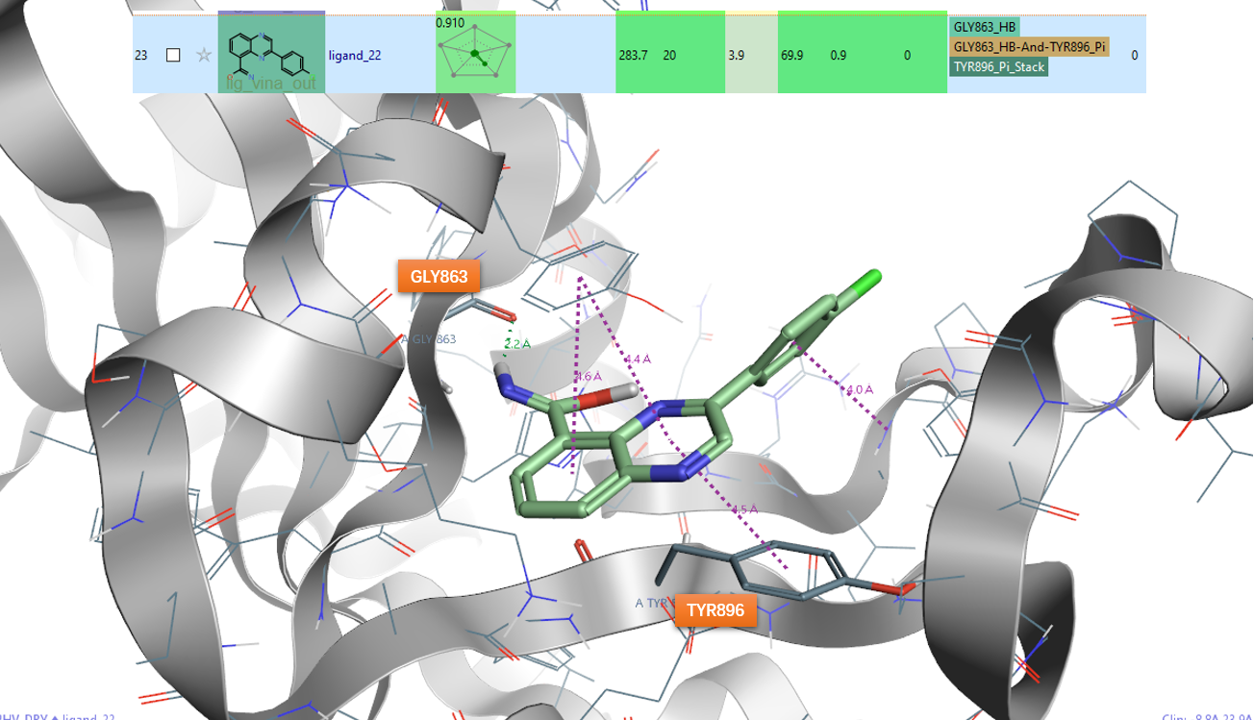
图4. 化合物ligand_22与靶标的相互作用分析,Python API的分析结果与视窗呈现一致,脚本可以代替人工分析过滤出与指定残基发生特定作用模式的化合物。
更详细的结果,请下载Flare项目文件用Flare Viewer来分析: liggrep_result.flr(分析完毕的文件)
4. 用RDKit的对Vina对接结果的氢进行优化
Flare内置了RDKit与OpenMM等软件,可以用RDKit的MMFF94力场进行优化计算。以其中一个分子为例,说明如何用RDKit提供MMFF94s力场对VINA的对接结果进行氢原子(重原子约束)的优化。
图5给出了优化前后的氨基氢取向比较,优化前NH2的氢基本与芳环垂直,这是不合理的;优化后NH2的氢与芳环共平面,这正是我们想要的。
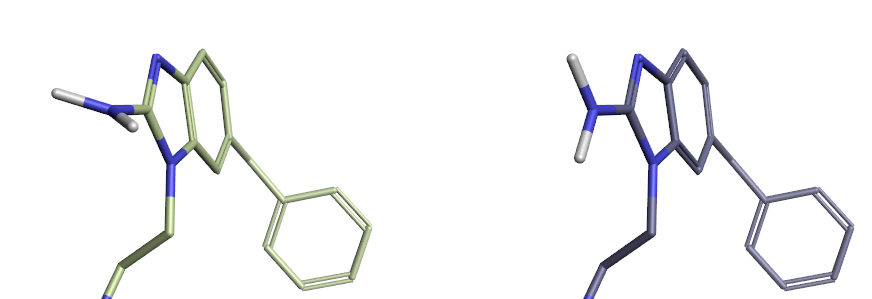
图5. Vina docking结果的氨基优化前后比较。左:Vina对接的结果;右:在RDKit中用MMFF94s对氢优化后的结果。
我相信在上述例子的提示下,用户可以修改上述代码,用一个循环实现对所有的分子进行优化。不过要注意的是,进行MMFF力场优化前需要是完整的分子,尤其不要忘记加上氢原子。
相关的应用场景
5.1 与指定残基发生相互作用配体的过滤
用来计算配体与特定残基之间的最短距离,实现将与某个残基相互作用的配体过滤出来。
5.2 用于共价抑制剂虚拟筛选
用于计算Michael受体与指定Cys-SH键距离以过滤出可能发生共价加成反应的pose,实现用常规分子对接进行共价抑制剂的虚拟筛选。
5.3 比较同一蛋白与不同配体在结合模式上的差异,理解SAR
距离的测量还可以用来自动分析、比较结合到同一结合位点不同配体的结合模式差异。以PDE5抑制剂Virgra与Cialis的共晶结构PDB 1UDT与1UDU为例,如图6所示,我们可以列出Virgra与Cialis同时发生相互作用的残基,呈现为金黄色的CPK;将仅与各自配体相互作用的残基按各自配体颜色呈现CPK。这样可以便利地发现两者在结合模式上的差异,快速发现关键的残基。
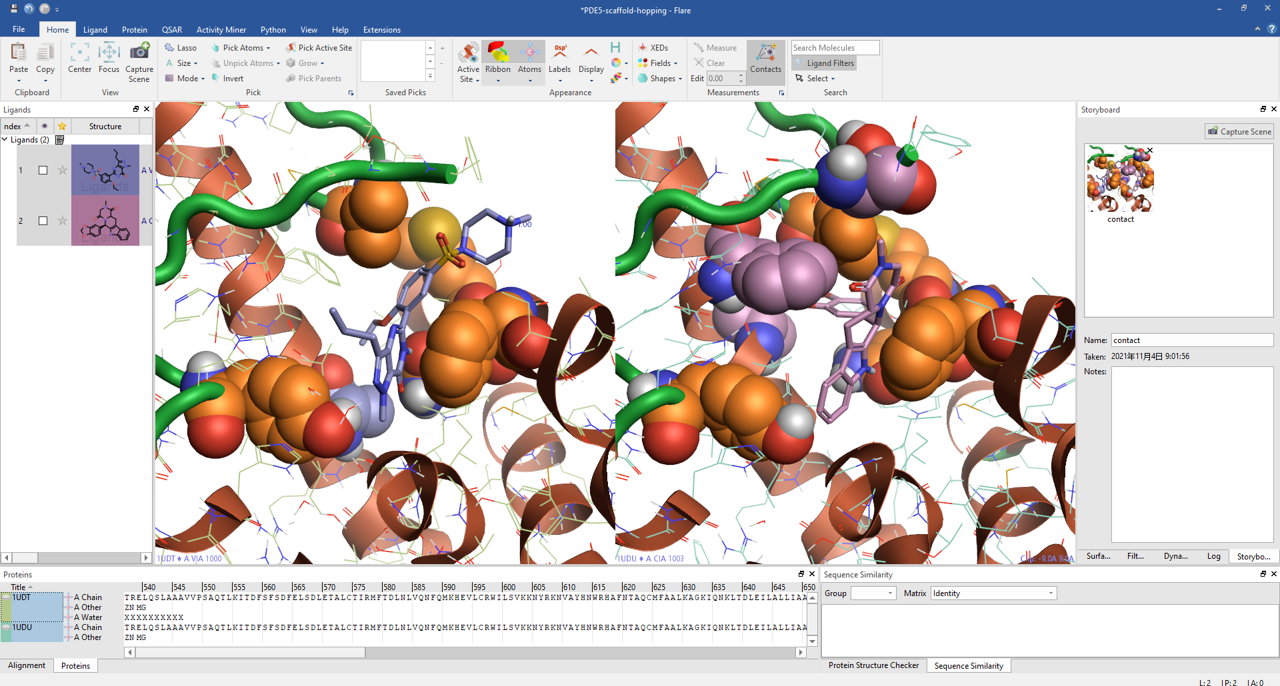
图6. 定义如果配体原子与残基原子间距小于等于3.5埃,则认为该配体原子与残基原子有相互作用。如果一个蛋白原子同时与Viagra、Cialis发生相互作用,则将该蛋白原子呈现为金黄色的CPK;如果一个蛋白原子仅与共晶的配体发生相互作用,则呈现为CPK,并按共晶配体颜色来着色。
还可以让python打印出具体的相互作用残基的共同与不同:

显然,3D图形显示比单纯的文本信息丰富多了:3D图形可以直观的体验在结合模式上的差异。这个比较过程如果单纯通过图形界面用鼠标操作将非常繁琐,而通过python的API调用则可一键完成。此外,python脚本还可以用于其它项目,一劳永逸。
更重要的是拓展到整个蛋白-配体数据库,可以回答如下的类似问题:在PDB BIND数据库中,与配体F原子相互作用的都是些什么样的蛋白原子与残基?这可以让你的计算团队快速采集数据、分析PDB数据库、统计相互作用模式,高效地为药化团队提供分子设计的决策依据。
6. 致谢
感谢中国海洋大学徐锡明博士[12]的指点,他指出Vina对接结果极性氢取向的问题。他是分子对接软件WATVINA的开发者,WATVINA是VINA的一个分支,支持药效团约束、水分子去溶剂化自由能贡献。
7. 参考文献
- Fischer, A.; Smieško, M.; Sellner, M.; A. Lill, M. Decision Making in Structure-Based Drug Discovery: Visual Inspection of Docking Results. J. Med. Chem. 2021, 64 (5), 2489–2500. https://doi.org/10.1021/acs.jmedchem.0c02227.
- Stahl, M.; Bo, H. Development of Filter Functions for Protein–Ligand Docking. J. Mol. Graph. Model. 1999, 3263 (98), 121–132. https://doi.org/10.1016/S1093-3263(98)00018-7.
- Beatriz, A. Chapter 14 CARTESIAN GENETIC PROGRAMMING AND THE POST DOCKING FILTERING PROBLEM. In Genetic Programming Theory and Practice II; O’Reilly, U.-M., Yu, T., Riolo, R., Worzel, B., Eds.; springer, 2005; pp 225–244. https://doi.org/10.1007/0-387-23254-0_14.
- Muthas, D.; Sabnis, Y. A.; Lundborg, M.; Karle, A. Is It Possible to Increase Hit Rates in Structure-Based Virtual Screening by Pharmacophore Filtering ? An Investigation of the Advantages and Pitfalls of Post-Filtering. J. Mol. Graph. Model. 2008, 26 (8), 1237–1251. https://doi.org/10.1016/j.jmgm.2007.11.005.
- Novikov, F. N.; Stroylov, V. S. Improving Performance of Docking-Based Virtual Screening by Structural Filtration. J. Mol. Model. 2010, 16 (7), 1223–1230. https://doi.org/10.1007/s00894-009-0633-8.
- Planesas, J. M.; Claramunt, R. M.; Teixidó, J.; Borrell, J. I.; Pérez-Nueno, V. I. Improving VEGFR-2 Docking-Based Screening by Pharmacophore Postfiltering and Similarity Search Postprocessing. J. Chem. Inf. Model. 2011, 51 (4), 777–787. https://doi.org/10.1021/ci1002763.
- Anighoro, A. Binding Mode Similarity Measures for Ranking of Docking Poses : A Case Study on the Adenosine A 2A Receptor. J. Comput. Aided. Mol. Des. 2016, 30 (6), 447–456. https://doi.org/10.1007/s10822-016-9918-z.
- Online, V. A. RSC Advances Computational Study of the Binding Orientation and affinity of PPARg Agonists : Inclusion of Ligand-Induced Fit by Cross-Docking. RCS Adv. 2016, 6 (69), 64756–64768. https://doi.org/10.1039/C6RA12084A.
- Ram, D. Is It Reliable to Take the Molecular Docking Top Scoring Position as the Best Solution without Considering Available Structural Data? Molecules 2018, 23 (5), 1038. https://doi.org/10.3390/molecules23051038.
- Ha, E. J.; Lwin, C. T.; Durrant, J. D. LigGrep: A Tool for Filtering Docked Poses to Improve Virtual-Screening Hit Rates. J. Cheminform. 2020, 12 (1), 1–12. https://doi.org/10.1186/s13321-020-00471-2.
- Gushchina, I. V.; Polenova, A. M.; Suplatov, D. A.; Švedas, V. K.; Nilov, D. K. VsFilt: A Tool to Improve Virtual Screening by Structural Filtration of Docking Poses. J. Chem. Inf. Model. 2020, 60 (8), 3692–3696. https://doi.org/10.1021/acs.jcim.0c00303.
- 在与中国海洋大学徐锡明博士的讨论中,他指出Vina对极性氢的处理方式以及对接结果氢的问题。他是分子对接软件WATVINA的开发者,WATVINA是VINA的一个分支,支持药效团约束、水分子去溶剂化自由能贡献。


