
用常规分子对接进行共价抑制剂的虚拟筛选
摘要:本文演示了如何用受体立体碰撞消除(SCAR)法进行共价抑制剂的分子对接虚拟筛选:将常规分子对接虚拟筛选与Ligandscout药效团打分组合,可以高效地实现该过程。JAK3算例表明:Ligandscout药效团打分与Fred分子对接组合,可以完美地将活性与非活性化合物区分开来;LIgandscout与Surflex-Dock组合也有相似的效果;将Fred或Surflex-Dock与Michael受体-残基结合点距离组合也可以实现共价抑制剂的显著富集。
肖高铿/2020-03-22
共价抑制剂的虚拟筛选
随着人们对共价抑制剂的发现越来越重视,越来越多的软件开始支持共价抑制的虚拟筛选。目前共价抑制剂虚拟筛选策略有分子对接虚拟筛选与药效团虚拟筛选。常用的共价对接软件有AutoDock4, CovDock, FITTED, GOLD, ICM-Pro与MOE[1]。采用药效团方法进行共价抑制剂虚拟筛选的工具不多,LigandScout是目前最常用的一个[2,3],它通过引入残基成键点(Residue binding point)的药效团元素(Feature)来实现虚拟筛选。此前的一个算例演示了如何用Ligandscout的这个特性进行EGFR激酶共价抑制剂的虚拟筛选[4]。
共价抑制剂的分子对接虚拟筛选
包括CovDock在内的共价抑制剂对接虚拟筛选商业软件,大都采用受体立体碰撞消除法(SCAR)[5],操作步骤包括[6,7,8]:1)将蛋白共价结合的残基突变为ALA;2)用常规方法将分子对接到突变后的蛋白结合位点(此时可以加以位置限制);3)选择pose;4)将docking pose的亲电基团与残基链成键;5)对复合物结构优化;6)打分与排序。其中步骤1、2与3是进行共价对接虚拟筛选的关键,而步骤4、5、6是用来预测结合模式并对pose排序。本文的目的是虚拟筛选,只需要完成前三步即可,因此本文将采用OpenEye/Fred等分子对接联合Ligandscout药效团识别来实现步骤1,2,3的过程,基本原理如下:
- 常规分子对接虚拟筛选
- Ligandscout药效团打分
- Michael受体与共价药效团特征间的距离
首先用OpenEye/Fred等分子对接进行虚拟筛选,将化合物对接到蛋白结合位点,每个化合物给出多个pose。
Ligandscout的药效团支持共价键的识别(图1),并支持对不同的药效团元素/特征加权重。Ligandscout不仅可以用来虚拟筛选,还可以用到作为打分函数:仅评估化合物的pose与药效团模型的匹配性。如果将共价药效团特征加权重,那么在打分时会优先将与共价药效团特征的pose给予高的分值。
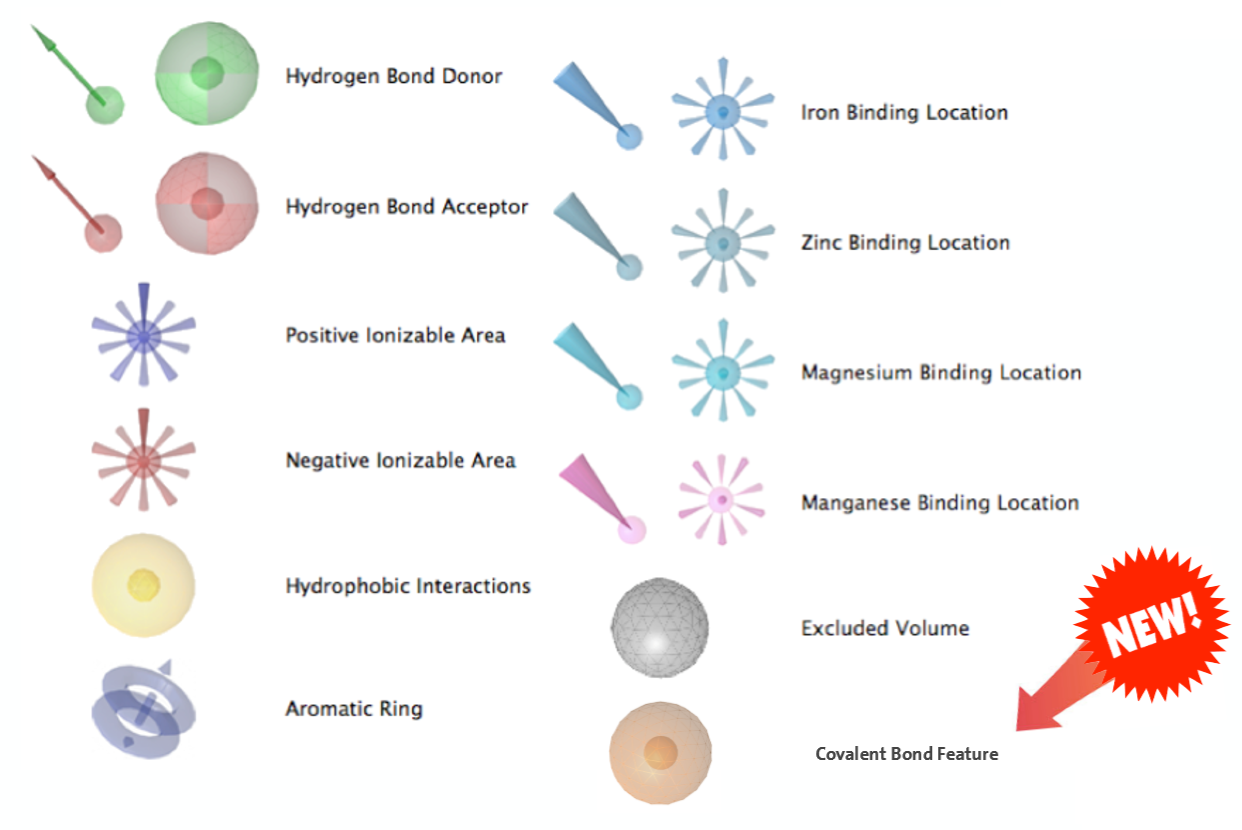
图1. Ligandscout的药效团元素/特征
用Ligandscout对对接过的配体进行药效团打分,可以筛选出识别出哪些pose与共价药效团特征匹配,哪些pose不匹配。那些Michael受体与Cys靠近的pose所代表的化合物,是潜在的共价抑制剂。富集这样化合物的过程就是实现共价抑制剂虚拟筛选的过程
当然,Michael与Cys巯基的靠近程度还可以直接用Michael受体与共价药效团特征间的距离来评估,并将该距离作为判断一个docking pose的Michael受体是否具备与Cys发生反应的前提条件。在文中的对接计算结果文件中,我们也给出这个距离,与药效团打分一起使用可以更好的选出符合条件的pose。
算例介绍:JAK3共价抑制剂的分子对接虚拟筛选
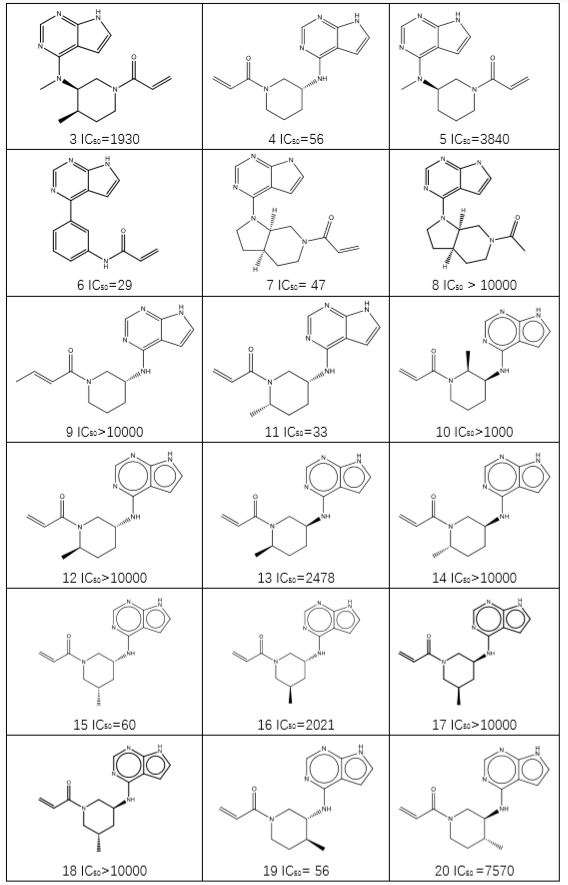
图 2. 辉瑞公司公开的18个化合物在底物[ATP]=1mM时的IC50(nM)[9]
PF-06651600(图2化合物11)是处于临床三期用于治疗斑秃的JAK3共价抑制剂,它通过Michael受体丙烯酰胺与CYS909共价结合实现JAK3对JAK1、JAK2与TYK的靶标选择性。PF-06651600的18个同系物活性差异巨大(图2)[9]:有的显示出JAK3的抑制剂活性,有的没有抑制剂活性。我们用SCAR策略,进行了虚拟筛选:1)从化合物11与JAK3的复合物结构蛋白(PDB 5TOZ)出发,将CYS909突变为ALA;2)将18个化合物对接到结合位点,每个分子给出20个pose;3)用Ligandscout进行药效团打分与结合亲和力打分,同时观察Michael受体与CYS909巯基的是否足够靠近;4)用下面方式对化合物进行打分:
- Ligandscout药效团打分(Unaligned Pharm. Score)
- Ligandscout结合亲和力打分(Binding Affinity Score)
- Michael受体的双键与CYS909巯基共价结合点的距离(COVALENT_DIST)
用来判断docking的pose能否与Cys909成共价相互作用,以及其它的相互作用,实现对共价抑制剂pose的挑选。
用来判断docking的pose与JAK3的结合亲和力,判断是否配体。对于类药的binder来说,该打分应该优于-25。
通过对接pose的Michael受体与Cys909的距离来初步判断Cys909是否有机会捕捉Michael受体,以实现对共价抑制剂的挑选。
本文将考察各种打分或其组合是否具备区分活性与非活性化合物的能力。
操作步骤
蛋白结构准备
Flare用来进行蛋白结构的准备,从下载PDB 5TOZ开始,具体方法参照《Flare 教程 | 分子对接-结合模式预测》[10]一文的第三节第1小节。
蛋白的突变
准备完毕,Flare的Protein表单有两个蛋白,一个是原始的5TOZ,一个是准备好的5TOZ_P;Ligand表单有配体A 7H4 0,单击该配体配,点击Home | Fit to window,则结合位点被居中、放大展示。注意:该配体是从共价状态剥离出来,Michael受体的不饱和双键被饱和了,可以通过编辑器将C-C单键改为C=C双键。
在3D视窗里用鼠标双击与配体共价的Cys909,其标记是配体丙烯酰胺烯烃的附近有个Cys的巯基,双击巯基选择该残基(图3,步骤1)。
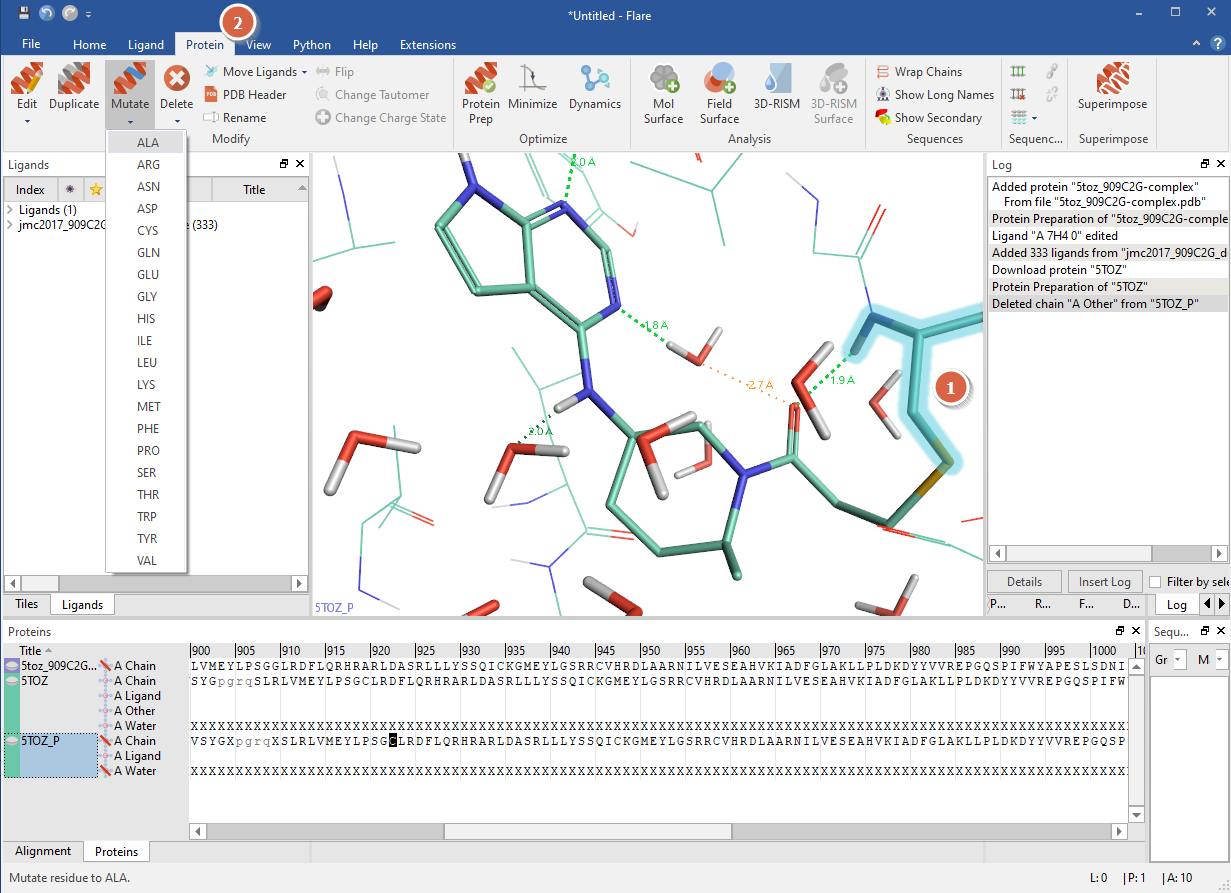
图3. 蛋白的CYS909的选择与突变设置
点击Flare | Protein | Mutate 菜单,选择下拉菜单里的ALA(图3,步骤2),出现图4的Flare Protein Preparation对话框,点击Start按钮,进行残基909的CYS2ALA突变。
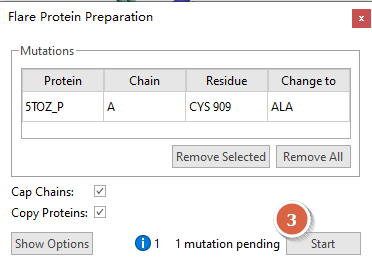
图4. 蛋白残基909C2A的突变
准备完毕,在蛋白表单区新增加了一个突变过后的蛋白5TOZ_P_X,从3D视窗与可以清楚的看到该突变。
导出蛋白与配体
在蛋白表单区选中other与Water链,右键|delete对之进行删除;然后在蛋白表单区用右键点击突变后的蛋白,选择export,保存为5toz_909C2G.pdb(注意:该下载链接的文件将CYS突变为GLY,而不是ALA)。同样,在配体表单区导出复合物结构里的配体,保存为reference.mol2以备后用。
同时也用OpenEye/OEDocking的Makereceptor准备了适合于OEDocking使用的受体文件:5toz_receptor.oeb.gz
配体分子的准备
从Thorarensen等人文章[9]的附件获得配体的SMILES代码,用RDKIT生成3D结构,保存为SDF文件:jmc2017.sdf。同时也用OpenEye/Omega准备了适合于OEDocking使用的构象数据库文件:jmc2017.oeb.gz
分子对接
可以采用任意的分子对接方法,比如Flare, Ligandscout/AutoDock Vina, OpenEye/Fred, Surflex Dock以及GOLD等等。在本算例中,我们采用了Surflex-Dock与OpenEye/Fred, 其中Surflex-Dock每个分子保存20个pose(对接结果:jmc2017_docked.sdf);OpenEye/Fred每个化合物保存50个pose(对接结果:jmc2017_fred.sdf)。
Ligandscout药效团打分与结合亲和力打分
用Ligandscout基于结构的方法从PDB 5toz生成药效团模型(图5),不考虑水分子的相互作用,该模型包含:1)铰链区的氢键与疏水相互作用;2)与Michael受体部分的氢键相互作用;3)以及共价相互作用(残基结合点)。
图5. 药效团模型:红色-氢键受体;绿色-氢键供体;黄色-疏水中心;橘黄色-残基结合点(共价结合点)。橘黄色药效团元素x2表示增加权重,以便共价抑制剂的打分更高、更容易被富集出来。
Ligandscout的药效团打分在对接的sdf格式结果文件里表示为Unaligned Pharm. Score,意思是docking的pose与该药效团的匹配程度:匹配程度越高,打分越高。
Ligandscout的Binding Affinity Score采用了Hyde score算法,对于类药化合物而言,binder的打分值应该优于-25[11],刘吉元等人[12]也成功地用该打分函数用于虚拟筛选发现先导化合物。Ligandscout的Binding Affinity Score在对接的sdf格式结果文件里表示为Binding Affinity Score。
Michael受体C=C双键到残基结合点的距离
为了更直观的考察docking获得的pose里的Michael是否靠近Cys909,还给出了Michael受体C=C中心到药效团残基结合点(见图5橘黄色药效团元素)的距离COVALENT_DIST,该值也包含在对接结果文件里。
结果
1. OpenEye/Fred分子对接
对接与打分的结果文件:jmc2017_fred.sdf。
Ligandscout药效团打分(大于45)与Fred的对接打分(FredScore小于-10kcal/mol)组合可以将活性好的化合物富集出来,命中化合物:4、5、6、7、11、15、16与19。这些化合物与909C2G的结合模式以及与药效团的匹配情况总结于图6。该组合打分几乎完美:将全部的高活性化合物包括在内,而将无活性或低活性化合物完全排除在外。读者可以自行下载Fred对接结果进行更多的考察。
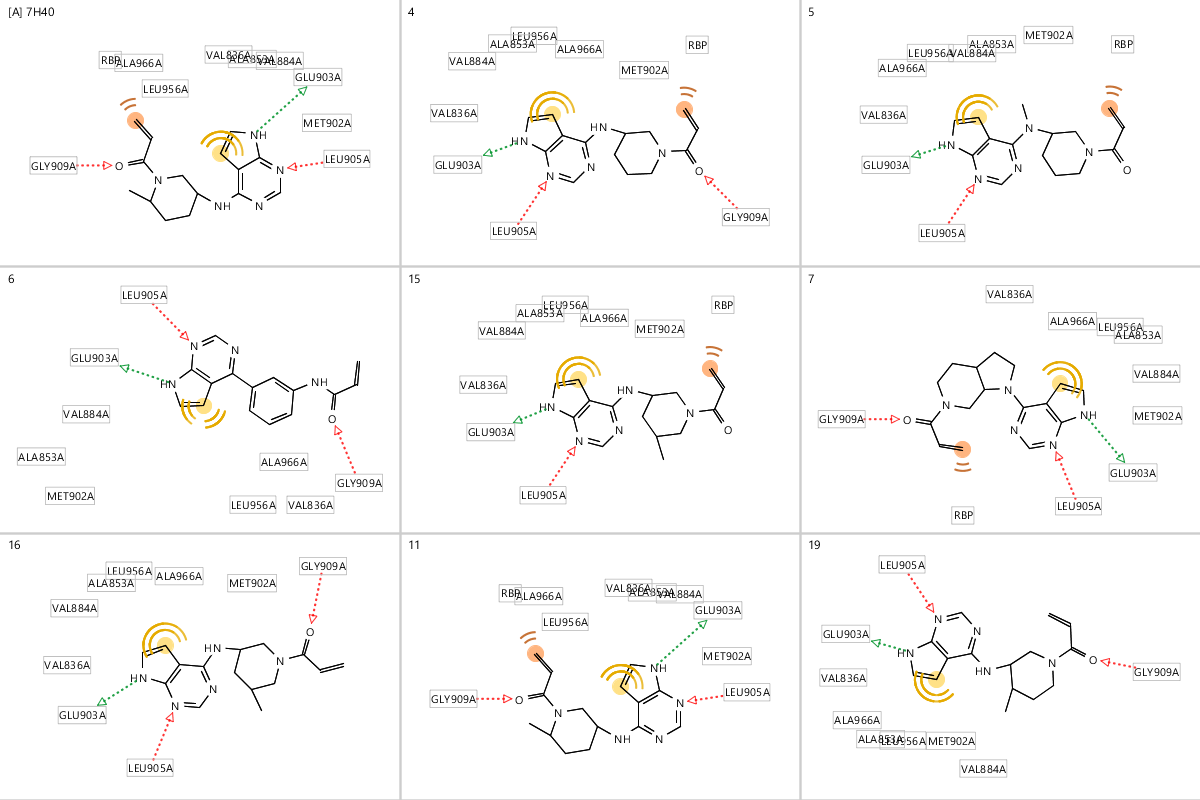
图6. Fred分子对接(小于-10)与Ligandscout药效团打分(大于45)命中化合物的结合模式
观察化合物5的50个对接结合模式会发现:FredScore(ChemGauss4)打分很好,但是Michael受体与Cys的距离既有小于1.5也有大于7,可视化分析发现这对应于Michael受体的两个空间取向。Pfizer的研究也表明:正因为对加成反应不利的构象占据了大约50的构象空间,所以化合物5对JAK3的活性也低。这说明,需要进一步对命中的化合物进行基于QM的构象分析(比如两面角扫描),以确保结合构象的稳定性。更详细的内容参见:Gaussian 算例 | 势能面扫描在Torsion Profile中的应用
FlareView与Ligandscout可以用来进行pose挑选,以Ligandscout为例,如图7所示,只要在配体表单区的Filter里设置过滤条件即可。

图7. 将docking结果读入Liganscout(File | insert),使用Filter功能,可以非常方便地根据分值挑选pose
总的来说,Fred分子对接与Ligandscout药效团打分组合可以实现共价抑制剂的虚拟筛选,将JAK3共价抑制剂活性化合物与非活性化合物区分地非常完美。
2. Surflex-Dock分子对接
对接与打分的结果文件:jmc2017_docked.sdf。
在对Surflex-dock对接结果进行Ligandscout药效团打分时没有对共价结合药效团元素添加权重,发现药效团打分就有足够的共价抑制剂富集能力。药效团打分大于45的化合物命中了化合物3、4、5、6、7、9、11、12、15、16与19。其中,除了化合物9与12没有活性之外,其它化合物都是活性化合物,包含了全部的高活性化合物。进一步将Binding Affinity Score大于-25的化合物去除,除了化合物3之外的高活性化合物均被命中。这说明,将Surflex分子对接虚拟筛选与Ligandscout药效团打分与Binding Affinity Score组合可以用来挑选有潜力的共价抑制剂。
3. Michael受体到共价结合点距离
如果一个docking pose的Michael受体C=C到共价结合点距离小于1.5[13],就认为该pose有可能发生共价相互作用。用这个距离可以非常方便地将潜在的共价结合pose富集出来。以1.5为截断值,在Surflex-Dock对接计算的结果里,命中化合物3、4、5、6、7、9、11、12、15、16与19,除了化合物9与12之外其他都是活性化合物;Fred Dock计算对接结果里,命中化合物3、4、5、6、7、9、10、11、12、15、16、17、18与19,其中化合物9、10、12与18没有活性,其余9个都是活性化合物,具有很高的命中率。这说明,用Michael受体C=C到共价结合点的距离也可以直接用来挑选合理的pose的。
与药效团打分比起来,距离打分给出的pose不如药效团打分的好看,主要体现在:药效团打分高的化合物,在铰链区也匹配的很好;而Michael受体到共价结合点距离近的pose不一定在铰链区也匹配的好。
筛选新的数据库
在《KNIME教程|基于反应的组合库生成》一文中我们演示了如何基于反应构建组合库,生成一个包含185个潜在的JAK3抑制剂的化合物库:jak3-results.smi。现在我们用上述流程来对该化合物库进行虚拟筛选:
- 化合物库的准备
- 分子对接虚拟筛选
- LigandScout药效团打分与距离打分
1 | omega2 -in jak3-results.smi -out jak3_lib.oeb.gz -enumNitrogen true -enumRing true -maxconf 1200 |
1 | fred -receptor 5roz_receptor.oeb.gz -dbase jak3_lib.oeb.gz -docked_molecule_file docked.sdf -num_poses 50 -save_component_scores true -prefix fred |
上一步docking的结果进一步用LigandScout药效团打分,并计算Michael 受体与Cys909的距离[13],结果见:jak3lib_scored.sdf。将Fred对接打分小于-10kcal/mol与距离小于1.5A的化合物过滤出来,仅保留Fred的docking打分最佳的一个,得到37个化合物:jak3lib_hits.sdf。
表1. 37个命中的化合物结构
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
接下来可以做什么
受体立体碰撞消除法分子对接将化合物在蛋白结合位点里摆pose,LigandScout将那些可以满足共价结合的pose挑了出来(当然不止于此),命中的化合物是潜在的共价抑制剂。这些化合物需要进一步过滤,可以考虑如下方面:
- 构象稳定系分析
- Cys909与Michael反应性的模拟
- Michael受体的遗传毒性模拟
如《选择性JAK3抑制剂PF-06651600的设计》一文所述,化合物的Michael受体在结合位点里有两个可能的取向:只有Michael受体朝向Cys909的构象为优势构象的化合物才是高活性化合物。因此必要进一步研究化合物的结合构象稳定性。比如化合物5,具有两个潜在的构象,他就比其类似物化合物4的活性差了很多。我们在《Gaussian 算例 | 势能面扫描在Torsion Profile中的应用》一文中对此单独进行了讨论。
不可逆的蛋白结合过程分两步:(1)首先形成可逆的初始非共价复合物EI;(2)初始的复合物EI进一步过渡为共价键结合的复合物E-I:
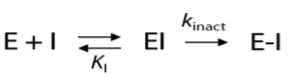
抑制剂的药效用二级速率常数kinact/Ki来表示。kinact反映了共价键的形成速率,而Ki反映了通过非共价相互作用形成初始复合物EI的稳定性。共价药物设计的目标是:发现结合过程由ki而不是kinact驱动的共价结合片段。第一步过程可以用基于结构的方法(比如分子对接)进行模拟;第二步需要涉及键的生成与断裂,需要用QM的方法进行模拟。在《可逆Michael受体硫醇加成反应的DFT理论计算》一文中分享了Houk课题组如何用Gaussian模拟Michael受体与Cys的反应性,并由此判断是否可逆或不可逆结合。
除了通过上述用模型分子粗略估算能垒,获得反应速率之外,还可以通过QM/MM计算获得反应自由能并精确估算反应速率,我们提供这样的计算服务与培训。往期的培训内容见:用QM/MM模拟酶与底物的反应。
此外,我们将提供一篇教程,通过模型分子讲解如何通过反应自由能计算获得反应速率,请持续关注我们的博客。
Michael受体可能脱靶与体内其它的亲核基团(比如DNA的碱基上的胺)反应引起毒性,所以共价抑制剂遗传毒性的评估是非常重要的。在《Gaussian教程 | 用DFT预测Michael受体的遗传毒性(AMES test)》分享了Grayson等人用Gaussian软件预测AMES毒性的方法。
文献
- Scarpino, A.; Ferenczy, G. G.; Keserű, G. M. Comparative Evaluation of Covalent Docking Tools. J. Chem. Inf. Model. 2018, 58 (7), 1441–1458. https://doi.org/10.1021/acs.jcim.8b00228.
- What’s new in Ligandscout 4.4. https://www.inteligand.com/download/Whats_New_in_LigandScout_4.4.pdf
- Schulz, R., Atef, A., et al. (2018). Phenylthiomethyl Ketone-Based Fragments Show Selective and Irreversible Inhibition of Enteroviral 3C Proteases. Journal of Medicinal Chemistry, 61(3), 1218–1230. https://doi.org/10.1021/acs.jmedchem.7b01440
- 肖高铿. Ligandscout教程 | 共价键药效团模型与虚拟筛选. http://blog.molcalx.com.cn/2018/05/15/ligandscout-tutorial-covalent-vs.html
- Ai, Y.; Yu, L.; Tan, X.; Chai, X.; Liu, S. Discovery of Covalent Ligands via Noncovalent Docking by Dissecting Covalent Docking Based on a “Steric-Clashes Alleviating Receptor (SCAR)” Strategy. J. Chem. Inf. Model. 2016, 56 (8), 1563–1575. https://doi.org/10.1021/acs.jcim.6b00334.
- Toledo Warshaviak, D.; Golan, G.; Borrelli, K. W.; Zhu, K.; Kalid, O. Structure-Based Virtual Screening Approach for Discovery of Covalently Bound Ligands. J. Chem. Inf. Model. 2014, 54 (7), 1941–1950. https://doi.org/10.1021/ci500175r.
- Zhu, K.; Borrelli, K. W.; Greenwood, J. R.; Day, T.; Abel, R.; Farid, R. S.; Harder, E. Docking Covalent Inhibitors: A Parameter Free Approach To Pose Prediction and Scoring. J. Chem. Inf. Model. 2014, 54 (7), 1932–1940. https://doi.org/10.1021/ci500118s.
- Introducing CovDock for Covalent Docking. https://www.schrodinger.com/newsletters/introducing-covdock-covalent-docking (accessed Mar 21, 2020).
- Thorarensen, A.; E. Dowty, M.; Ellen Banker, M.; Juba, B.; Jussif, J.; Lin, T.; Vincent, F.; M. Czerwinski, R.; Casimiro-Garcia, A.; Unwalla, R.; et al. Design of a Janus Kinase 3 (JAK3) Specific Inhibitor 1-((2 S ,5 R )-5-((7 H -Pyrrolo[2,3- d ]Pyrimidin-4-Yl)Amino)-2-Methylpiperidin-1-Yl)Prop-2-En-1-One (PF-06651600) Allowing for the Interrogation of JAK3 Signaling in Humans. J. Med. Chem. 2017, 60 (5), 1971–1993. https://doi.org/10.1021/acs.jmedchem.6b01694.
- Flare 教程 | 分子对接-结合模式预测. http://blog.molcalx.com.cn/2017/06/04/flare-tutorial-dock-imatinib.html
- Reulecke, I.; Lange, G.; Albrecht, J.; Klein, R.; Rarey, M. Towards an Integrated Description of Hydrogen Bonding and Dehydration: Decreasing False Positives in Virtual Screening with the HYDE Scoring Function. ChemMedChem 2008, 3 (6), 885–897.
- Liu, J., et al. (2016). “Structure-based discovery of potentially active semiochemicals for Cydia pomonella (L.).” Scientific Reports 6: 34600. DOI:10.1038/srep34600
- 计算Michael受体与Cys-SH距离的方法参见:肖高铿. 在Flare里如何将与某个残基相互作用的配体过滤出来. http://blog.molcalx.com.cn/2019/03/28/pyflare-faq.html#flare_tag


