
摘要:大量致力于JAK激酶抑制剂的发现工作最终将几种化合物推进到临床开发并有两个被FDA批准。尽管在过去20年中付出了巨大的努力,但是高选择性的JAK3抑制剂一直未能找到。辉瑞公司的研究人员经过巨大的努力发现了首个口服活性的JAK3特异性抑制剂,其通过与JAK3特有的残基CYS-909共价相互作用实现JAK同工酶选择性抑制。JAK3酶共价抑制剂设计的难点在于其具有相对快速的再合成速率,因此要求该酶的共价抑制剂不仅具有合适的药动学性质(pharmacodynamics properties)而且还要将不希望的脱靶反应性限制到最小范围。本研究的努力最终发现了化合物11(PF-06651600),不仅具有体内活性而且清除率较低。鉴于化合物11的有利药效和安全性,该化合物进入临床研究评价阶段。
编译:肖高铿/2019-10-26
原文:Thorarensen, A.; et al. Design of a Janus Kinase 3 (JAK3) Specific Inhibitor 1-((2 S ,5 R )-5-((7 H -Pyrrolo[2,3- d ]Pyrimidin-4-Yl)Amino)-2-Methylpiperidin-1-Yl)Prop-2-En-1-One (PF-06651600) Allowing for the Interrogation of JAK3 Signaling in Humans. J. Med. Chem. 2017, 60 (5), 1971–1993. https://doi.org/10.1021/acs.jmedchem.6b01694.
JAK3选择性抑制剂
JAK激酶(JAK)是I/II型细胞因子受体信号转导所必需的非受体酪氨酸激酶。JAK家族有四个成员:JAK1、JAK2、JAK3和TYK2。JAK1、JAK2和TYK2普遍表达,而JAK3主要表达于白细胞中。所有的四种JAK同工酶都参与了免疫相关的功能,而JAK1和JAK2还在造血、生长和神经元功能等相关功能中的起着关键作用。JAK信号转导的显着特征是在细胞因子受体复合物中形成JAK激酶二聚体,细胞因子受体信号所需的JAK二聚体包括JAK1/JAK3,JAK1/JAK2,JAK1/TYK2,JAK2/ TYK2和JAK2/JAK2。
鉴于JAK家族成员在细胞因子信号转导中的重要性,促使人们投入大量精力去开发选择性JAK抑制剂1,2。部分抑制剂针对自身免疫性疾病、各种癌症和骨髓增生性疾已经进入临床研究阶段,迄今获得了两项FDA批准:托法替尼(Tofatinib,1)和鲁索替尼(Ruxolitinib,2)3。JAK3缺陷的人类SCID患者为JAK3作为靶标提供了强力的验证,并使得JAK3成为治疗性干预的第一批JAK靶标之一。尽管如此,迄今为止报道的大多数抑制剂在JAK家族中缺乏选择性。因此,JAK3选择性抑制剂的治疗潜力仍有待解决。事实上,人们对公共γ-链(common γ-chain)细胞因子信号转导中JAK1与JAK3所起的作用还存在争议4。最近,我们证明了选择性JAK3抑制剂能够在体外有效地抑制IL-15介导的信号传导,显示了选择性JAK3抑制的治疗潜力5。
|
|
|
| Tofacitinib,1 | Ruxolitinib,2 |
Figure 1. FDA批准的JAK抑制剂
四个JAK家族成员ATP结合位点的序列比对表明,JAK3仅具有两个可用于设计选择性抑制剂的独特残基3。此外,JAK亚型的晶体结构研究表明家族成员仅具有极其微小的构象差异6。除了结构生物学信息之外,再加上JAK3与其他家族成员相比具有更高的ATP亲和力,意味着高选择性、ATP竞争性的JAK3抑制剂设计将非常困难5。JAK3中特有的CYS-909残基提供了以共价键设计选择性抑制剂的机会,克服了ATP结合腔内的高序列相似性和形状相似性带来的选择性抑制剂设计挑战。这个思路已有文献报道,并获得体外实验数据支持7,8。以共价方式靶向JAK3为药物设计提供了又一个可选的方式,具有靶标失活后延长药效学(pharmacodynamics,PD)的潜力。共价修饰也具有其独特的负面问题,例如可能导致不良药理作用的脱靶反应性以及非传统的代谢清除率。我们最近公开了选择性共价JAK3抑制剂11(PF-06651600)并总结了多个炎症疾病临床前体内模型的药效学研究结果9。这个高度优化的共价JAK3抑制剂可以让激酶快速地失活且具有最小的化学反应性、良好的ADME性能和整体选择性。在本文中,我们描述了为发现化合物11所付出的努力。
共价抑制剂设计的灵感来源
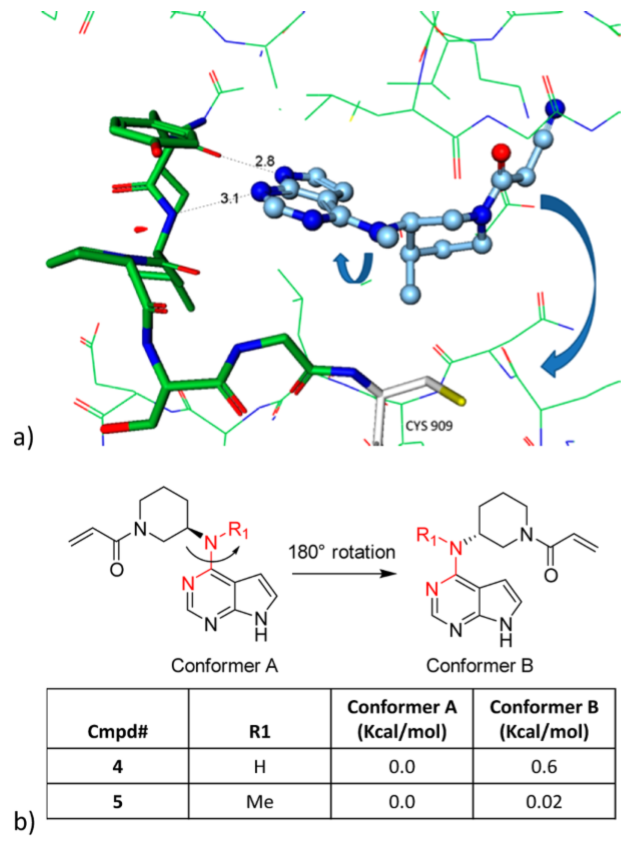
Figure 2. (a)Tofacitinib与JAK3复合物晶体结构提示围绕N-C轴旋转可以将酰胺放置在离CYS-909很近的位置。(b)N-Me对构象A的能量影响很大,化合物4的A与B构象的比例为70:30
最初的设计灵感来源于结构生物学,Tofacitinib与JAK3激酶结构域的复合物晶体结构为与CYS909共价结合的抑制剂设计提供了灵感10。围绕嘧啶胺N-C键旋转可能会使酰胺取代基紧邻CYS909,适当设计的亲电试剂可以捕获到亲核的半胱氨酸(图2a)。将丙烯酰胺放在托法替尼母核上得到化合物3。化合物3对JAK3的抑制作用中等,而对JAK家族的选择性较差,这表明可逆的(Ki)蛋白相互作用是结合亲和力(表1)主导力量。 X射线晶体结构分析支持了这一假设:发现吡咯并嘧啶铰链结合片段在活性位点排列整齐,而丙烯酰胺取代的哌啶高度无序(数据未显示)。由于JAK3在其家族成员中对ATP的亲和力最高,所以靶标抑制的Ki成分一定不能成为活性的主要驱动力,而只能通过与CYS909的共价相互作用来提供合适的选择性。
Flare视频演示:初始的灵感来源于旋转Tofacitinib的嘧啶N-C键,可使得酰胺基往CYS909靠近。这提示:适当设计的亲电基团可以捕捉到JAK3 CYS909上的巯基。
为了增加对JAK3的选择性,必须提高失活速率(inactivation rate, kinact)。采用DFT方法的模拟研究表明11 :去除N-甲基和哌啶顺式甲基后得到低能构象的衍生物,它们的亲电弹头(warhead)紧邻CYS909,见Figure 2b。事实上,化合物4表现出显著地活性提升(JAK3 IC50 = 56 nM,[ATP] = 1 mM)。在相对较长的测定反应时间条件下,IC50可以很好地衡量不可逆抑制剂的活性强度,因为它与时间无关的kinact/ Ki具有很好地相关性12,13。实际上,在基于细胞的体外和体内试验中,IC50易于测定、对数转化后可以体现细胞水平的体内、外功能,这使得IC50成为研究的首选参数。化合物4相对于3具有更高的JAK3选择性表明kinact参数已经得到显着改善。后续研究进一步证实化合物4与CYS909形成了共价键,并且化合物4的离解速率大于8h。
 |
 |
| a | b |
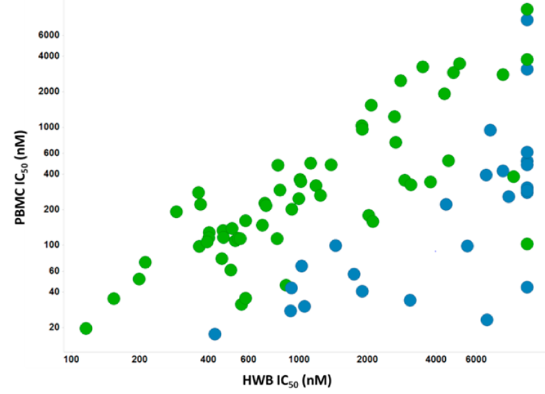
Figure 3. (a)IL-15 PBMC测试与对应的HWB测试之间的相关性。苯胺基丙烯酰胺=蓝色,脂族丙烯酰胺=绿色。(b)HWB测试中化合物稳定性:T1/2:小于100分钟,品红色; 100-200分钟,棕褐色; 200-300分钟,黄色; 大于300分钟,绿色(n = 107)。
利用共价“手柄”的高JAK3特异性的概念可以拓展到一系列亲电试剂和模组(motif),这些亲电试剂和模组将亲电试剂放置在CYS909附近,这样的衍生物类如苯胺基丙烯酰胺化合物6,其显示出强的JAK3抑制(IC50=29nM)。化合物4和6代表了平行探索的两个不同系列(即脂族和芳基氨基丙烯酰胺)。虽然每个系列的大多数化合物的活性是通过快速共价失活(即kinact)获得的,但每个系列都是通过明显不同的原理实现的。用谷胱甘肽(GSH)作为捕集剂进行的亲电性定量实验表明,这两个系列具有非常不同的固有化学反应性(芳基氨基丙烯酰胺,T1/2=0.4-4h); 脂肪族氨基丙烯酰胺(4-40 h)14。芳基氨基丙烯酰胺已成功地用于JAK3、EGFR和BTK等靶标的不可逆抑制剂的设计, 目前尚不清楚这些不同的相对反应性会产生何等程度的脱靶效应。当活性测试从基于细胞的PBMC测试转为人全血(富含蛋白质的培养基)的测试时,反应性的影响变得相当明显(图3a)。脂肪族氨基丙烯酰胺在不同的测试之间表现出紧密的相关性,而芳基氨基丙烯酰胺相关性较差。脂肪族氨基丙烯酰胺与芳基氨基丙烯酰胺之间存在着这种转化差异的原因尚未完全阐明; 然而,上述芳基氨基丙烯酰胺的强反应性可能是主要原因。此外,由两个系列代表性化合物进行的血液稳定性研究显示出相似的趋势(图3b)。因此,在基于酶和PBMC细胞的测试中,尽管芳基氨基丙烯酰胺类化合物产生了最有效的衍生物,但很明显,该系列化合物固有的化学反应性水平需要被减弱以便在全血环境中具有最佳的活性。
口服活性JAK3特异性抑制剂的关键参数
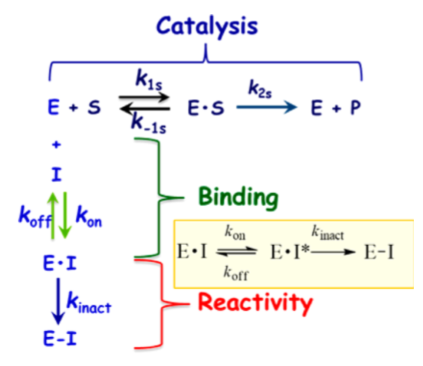
Figure 4.JAK3 CYS909共价修饰过程中的各个结合与反应事件
化合物4的发现阐明了用于将来分子设计至关重要的指导原则。衍生物必须有足够的Ki以便高效地与ATP竞争并与引发与蛋白的结合,但这只是酶抑制剂的一小部分。快速地失活(kinact)依然是优化的主要驱动力,因为它是获得足够选择性的决定步骤。从上述关于芳基氨基丙烯酰胺的讨论结果可以认为:简单地提高化学反应性kinact不是富有成效的优化策略。当考虑共价加合物形成中涉及的各个事件时,初始的结合复合物EI不一定代表反应性的复合物EI*(图4)。因此,增加可逆结合的EI*复合物的浓度应该可以加快共价捕获。以化合物6为代表的系列化合物,其强效化合物所需的化学反应性增加可能表明存在低浓度的EI*复合物15。增加EI*复合物浓度的设计原理可以用化合物4来说明。化合物4和6与JAK3的复合物晶体结构为分子设计提供了重要指导,重点是反应构象的稳定化(图5a、b)。这两种化合物都与CYS909加成,但它们的构像有着明显不同的丙烯酰胺空间排列。对于4,观察到丙烯酰胺形成两个氢键:一个键合到与配体嘧啶氮相互作用的保守水分子上,第二个键合到CYS909酰胺的NH上。虽然这些氢键相互作用是在共价键形成之后观察到的,但我们的假设是它们也存在于EI*复合物中,稳定了反应性构象并活化丙烯酰胺以加成到CYS-909中。相反,6中的丙烯酰胺不保留任何这些相互作用,因此需要增加的固有化学反应性来捕获Cys-909。将这些见解应用于具有环约束的化合物7的设计。化合物7与Jak3的复合物X-射线晶体结构说明哌啶环环化回到连接臂(linker)胺限制了哌啶-N键周围的旋转,并使哌啶稳定在合适的椅式构象中,以提供用于Cys-909亲核攻击的丙烯酰胺,图5c。
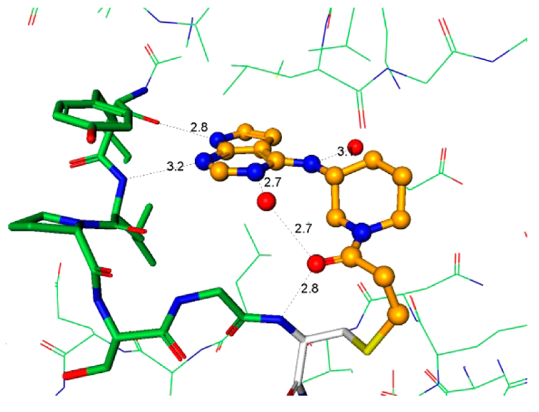 |
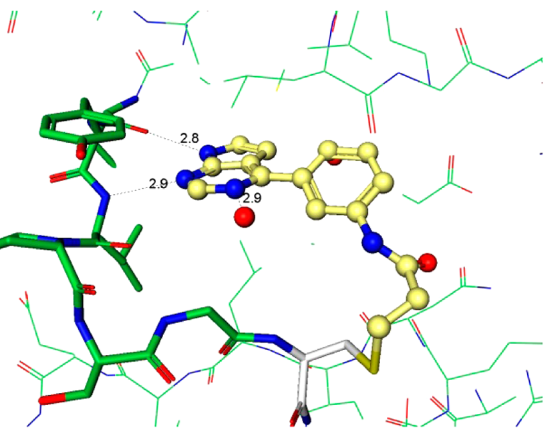 |
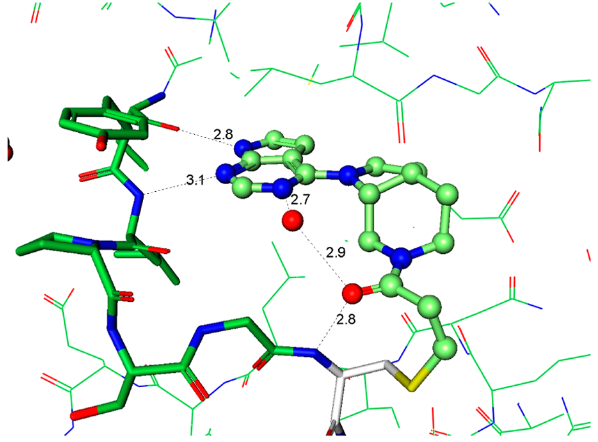 |
| a(4,PDB:5TTS) | b (6,PDB:5TTV) | c (7,PDB:5TTU) |
Figure 5. 化合物4、6、7与JAK3 CYS-909共价结合化合物的晶体结构
JAK1、JAK2和TYK2的再合成速率相当快,在2-4h的范围内16。缺乏JAK3的信息促使人们去测量PBMC细胞中的再合成速率,其被确定为3.5h17。这种相对快的再合成速率意味着由于共价抑制而带来的药效学(PD)效应延长的潜力超出了经验范围。这与诸如FAAH18、BTK19之类靶标的缓慢再合成速率的经验相反。 在这情况下,相对短期的药物暴露可在体内提供长期的靶标抑制。但是对于JAK3而言,需要具有低清除率的优异药代动力学性质(PK)以实现持续的靶标失活和高水平的药效。
表2. 共价JAK3抑制剂的ADEM性质
| Compd | HLM (μL min−1mg−1) | RLM (μL min−1mg−1) | RRCK (10−6cm/s) | Human blood T1/2(min) | Rat blood T1/2(min) |
|---|---|---|---|---|---|
| 4 | 8 | 小于15 | 8 | ~332 | 98 |
| 7 | 12.2 | 70 | 20 | 192 | 164 |
| 8 | 小于8 | 25 | 16 | NA | NA |
| 11 | 8 | 小于15 | 15 | 大于360 | 161 |
尽管肝微粒体判断的氧化稳定性非常合理(表2),但是化合物4(数据未显示)和7在大鼠中表现出从中等到差的PK性质。用化合物7和相应的乙酸酯衍生物8进行的一组PK实验提供了对药物清除机制的关键见解。对于这两种化合物,在大鼠体内的PK实验中可以观察到非常高的清除率(表3)。为了考察氧化代谢对总清除率的贡献,在进行实验之前先在用广谱的细胞色素P450(CYP450)抑制剂氨基苯并三唑(ABT)处理。结果发现乙酸酯8的清除率显著降低,而丙烯酰胺7仍被高度清除,这说明存在着非CYP450清除途径。事实上,谷胱甘肽-S-转移酶(GST)介导的谷胱甘肽(GSH)加成到丙烯酰胺上是造成这种差异的原因20。为了建立SAR模型以理解GST介导的清除,开发了全血稳定性测试方法以便体外方法测量全身GST介导的清除。该方法作为预测工具的有效性最近已获验证21。
表3. 在添加或不添加广谱CYP450抑制剂ABT的情况下化合物7和8在大鼠体内的药代清除率
| Compd 7 | Compd 8 | |
|---|---|---|
| CL (mL min−1kg−1),−ABT | 109 | 76 |
| CL (mL min−1kg−1),+ABT | 81 | 10 |
尽管化合物7的PK较差,但仍在体内研究中利用小鼠迟发型超敏反应(DTH)进行了评估,DTH是一种依赖于T细胞的临床前模型,可用于表征化合物7。在预防性小鼠DTH模型中,第0天通过皮下注射绵羊红细胞(sRBC)引发小鼠而引起了免疫应答,并在第5天向右后足垫注射了sRBC以引起可测量的炎症反应。 在整个研究中,化合物7每天给药一次。 7剂量依赖性地显著地抑制了右后足垫中的DTH反应,并且在每天两次低至3 mg / kg给药剂量时观察到疗效。 在该DTH模型中,化合物1每天两次每次30 mg / kg的给药剂量也是有效的(图6)。 这为我们提供了初步的支持,即选择性抑制JAK3可以产生相应的体内功能性。
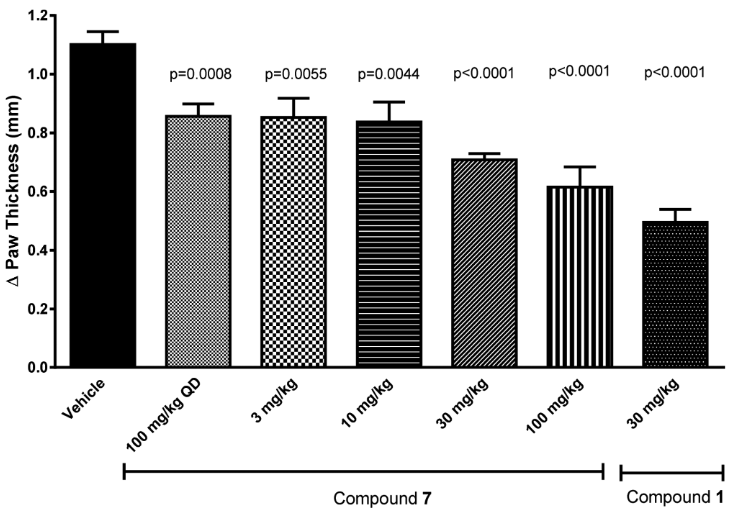
Figure 6. 化合物7的药效。8周龄雌性BALB/c小鼠(Taconic)在第0天通过皮下注射2×107绵羊红细胞(sRBC)致敏。在致敏前第0天开始口服给药持续进行至第6天。在右后脚垫上用1×108 sRBC攻击小鼠,左后脚垫接受无菌PBS注射。在注射之前测量爪的厚度以进行基线测量,然后在24h再次通过卡尺测量。与空白给药的小鼠相比,爪子厚度以剂量依赖性方式显着降低。 除另有说明外,所有化合物每天两次给药。
设计口服活性的JAK3专一性抑制剂
Table S1. PF-06651600对JAK家族的抑制活性
SEM:standard error of the mean; n: the number of independent experiments; IC50:the geometric mean of “n” independent experiments
关于调节GST介导清除的设计策略的知识非常有限,一般而言,GST是一类可耐受多种底物的酶22。因为亲电体周围的空间位阻会降低GST催化GSH添加的能力,所以在早期的时候我们试图在丙烯酰胺中添加取代基来抑制化学反应性。该方法显著地降低了GST介导的清除率,但不幸的是与JAK3亲和力不兼容。 例如,将β-甲基加到4的丙烯酰胺部分会产生几乎没有活性的类似物9(IC50大于1000nM)。一种替代方法是加入长程立体空间“手柄”以调节GST结合,氰酰胺系列JAK3抑制剂就使用了这样的策略23。尝试将该策略应用于类似物4,探索了在哌啶环不同位置进行甲基取代对活性的影响。关键的初始衍生物10(表5)相对于去甲基类似物4显示出增强的血液稳定性,但不幸的是,其对JAK3的活性较差。我们认为该化合物活性降低的原因是:化合物10与ATP结合口袋中的LEU956残基侧链发生空间冲突,导致捕获CSY-909所需的反应构象不稳定。我们经过模拟发现,将氨基吡咯并嘧啶基团从2位置移至4位置将导致与CSY-909相互作用的高度有利的基态构象,而不会与JAK3的ATP口袋中的任何残基发生空间碰撞。通过这种构象,我们预期丙烯酰胺羰基与CYS909的NH相互作用会进一步稳定反应性构象,并有可能激活丙烯酰胺进行亲核加成。这个设想在化合物11中得以实现,化合物11是一种JAK3高度特异性的抑制剂(IC50 = 33 nM),在1 mM ATP浓度下相对于其他JAK酶没有相关活性,X-射线晶体学也证明化合物与CYS909发生了共价结合23(PDB code:5TOZ)。体外高活性的化合物11在细胞(IL-15刺激的PBMC,IC50 = 51 nM)和人全血分析(IL-15刺激的HWB分析,IC50 = 197 nM)中均显示出色的活性(表1)。在将全血IC50转换为106 nM的游离IC50(在PMBC IC50的2倍以内)之后,化合物11驱动JAK3药理学的游离药物浓度的重要性是显而易见的。可以观察到化合物11对其他常见的γ链细胞因子信号转导的抑制作用,而对其他JAK激酶信号相关的细胞因子抑制作用却很少17。还观察到细胞水平的功能药效与JAK3酶与化合物11的结合率相关性很好24。重要的是,2-甲基片段对人肝微粒体、肝细胞和血液的稳定性非常重要。体内药代动力学研究表明,化合物11在大鼠和狗中的清除率较低,但在猴子中的清除率较快(相对于肝血流量)且口服生物利用度较低(表4)。
表4. 化合物11在不同种属中的体内、外PK性质
| Species CL Assay | Mouse | Rat | Monkey | Dog | Human |
|---|---|---|---|---|---|
| Hepatocyte CLint,u ((μl/min)/million cells) | 53 | 14 | 7.6 | 2.1 | 2.8 |
| Blood t1/2(min) | 大于360 | 161 | 37 | 大于360 | 大于360 |
| observed in vivo CLb(mL min-1kg-1) | 48 | 53 | 42 | 8.6 | – |
| Vss(L/kg) | 0.8 | 1.4 | 2.6 | 1.1 | 1.3b |
| in vivo plasma t/2(h) | 1.3 | 0.3 | 0.7 | 1.1 | 1.8c |
| po bioavailability(%) | 62 | 85 | 56 | 109 | – |
aCLint,u=unbound intrinsic clearance; CLb=blood clearance.;bPredicted from allometric scaling of unbound Vss;cPredicted human half-life
跨物种的药代动力学曲线与肝细胞和血液中的稳定性直接相关,这说明应用体外工具来预测体内药代动力学行为是有价值的。化合物11的动物(鼠/狗)口服生物利用度较高,这与其体外高被动通透性(RRCK,15 × 10-6cm/s)、高水溶性(大于2mg/mL)和低肝清除率相一致。化合物11的代谢清除是通过氧化代谢(丙烯酰胺和主要通过CYP3A4的环氧化)和谷胱甘肽共轭(在丙烯酰胺上)介导的。化合物11的大鼠肾脏和胆管清除率低。基于对清除机制和体内外相关性的理解,预测化合物11的人体血液清除率约为5.6mL min-1kg-1 18。同样预测人体口服生物利用度约为90%,半衰期约为2小时。这些结果促使我们对哌啶环其他位置甲基取代的影响进行了系统评估(表5)。
| ID | Compound | Jak3 1mM IC50 (nM) | HBlood Stab t1/2(min) |
|---|---|---|---|
| 10 |
|
大于1000 | 大于360 |
| 11 |
|
33 | 大于331 |
| 12 |
|
大于10000 | 185 |
| 13 |
|
大于2478 | 大于360 |
| 14 |
|
大于10000 | 大于360 |
| 15 |
|
60 | 大于250 |
| 16 |
|
2021 | 103 |
| 17 |
|
大于10000 | 304 |
| 18 |
|
大于10000 | 大于360 |
| 19 |
|
56 | 大于300 |
| 20 |
|
7570 | 142 |
在化合物4-JAK3复合物晶体结构中观察到优势的椅式构象环取代的衍生物通常将亲电试剂放置在CYS909附近并产生效果 (比如化合物15和19)。GST介导的清除率受到多方面的影响,比如对映异构体构型(化合物14 vs 12)和相对立体化学构型(化合物11 vs 12)对血液稳定性具有显著影响。这些实践使我们发现了衍生物19,它通过令人惊讶的远程空间效应而具有优异的活性 (IC50=56nM)和血液稳定性。
表6. 化合物11对其它激酶的失活率,在与JAK3相同的位置上这些激酶也具有CYS残基。
| Kinase | kinact(s-1) | Ki(μM) | kinact/Ki(M-1*s-1) | IC50(1 mM ATP)(nM) |
|---|---|---|---|---|
| JAK3 | 2.32 | 6.31 | 3.68×105 | 90 |
| BMX | 0.00564 | 0.543 | 1.03×104 | 606 |
| ITK | 0.000144 | 0.0269 | 5.35×103 | 8510 |
| TXK | 0.000487 | 0.131 | 3.79×103 | 193 |
| TEC | 0.00156 | 0.679 | 2.30×103 | 592 |
| BTK | 0.124 | 62.3 | 1.99×103 | 607 |
| BLK | 0.0278 | 32.4 | 8.58×102 | 19000 |
| HER4 | ND | ND | ND | 25200 |
| EGFR | ND | ND | ND | 大于50000 |
| HER2 | ND | ND | ND | 大于50000 |
| MAP2K7 | ND | ND | ND | 大于50000 |
IC50值在Carna Bioscience测定
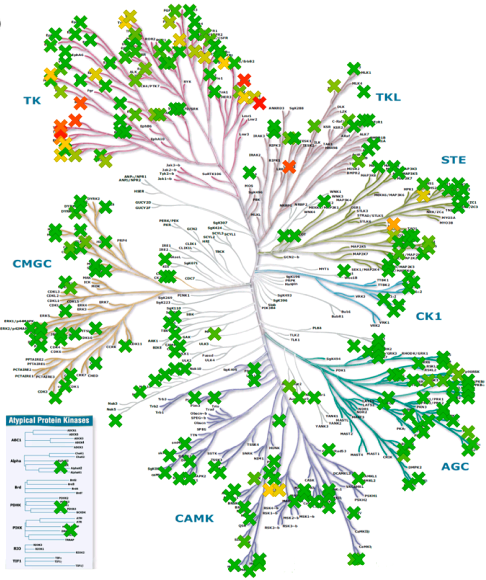
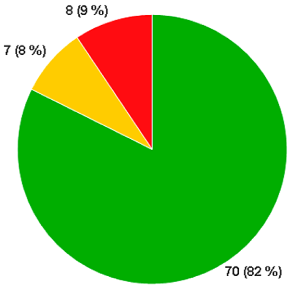
化合物11对305种激酶具有高选择性(Invitrogen,图7)。大约200种激酶在ATP结合位点中含有CYS残基26。在这组激酶中,11种激酶含有与JAK3相同位置的CYS。对该亚组激酶进行评价时,化合物11对8个靶标显示出可测量的活性(表6)。当底物ATP浓度为1mM时,这些靶标的IC50显示出相对于JAK3良好的选择性。还测定了化合物11对这些靶标的Ki和kinact贡献。有趣的是,kinact贡献是JAK3抑制的主要特征,这说明该化合物对亲电试剂的排列进行了最佳优化,并且通过CYS-909加成而使激酶快速失活。相反,ITK的失活非常慢,并且其大部分亲和力是由Ki贡献的。非常慢的失活速率需要可观的滞留时间用于在有高ATP水平的细胞环境中进行捕获,这表明化合物11是ITK的无效抑制剂。此外,当比较BMX和TXK时,使用严格的kinact/Ki作为选择性的量度忽略了与ATP的竞争对功能结果的不同影响。我们还观察到化合物11对其他三种ATP结合位点含CYS的激酶(SLK、FGR和FLT3)具有非常弱的亲和力, 并且这些激酶都没有以共价方式被抑制。
 |
 |
| a | b |
Figure 7. a(左). 化合物11在1μM时对Invitrogen公司305个激酶的激酶谱分析结果:颜色代表百分抑制剂:深绿色0%;黄色50%;深红色100%。选择性Gini系数=0.88。b(右). 测试过的、ATP结合位点含有CYS残基的激酶。
化合物11对体外细胞信号传导的有效抑制清楚地说明JAK3的选择性抑制可以通过利用异源二聚体JAK1/JAK3对共同的γ链细胞因子受体有效地调节信号传导。此外,在γ链信号驱动的多种体内炎性疾病模型(AIA、DTH、CIA和EAE)中报告的11和7的高度疗效明确地证明了选择性JAK3抑制多种人类疾病的治疗潜力17。在这些结果的基础上,选择化合物11进一步用于临床评价。
结论
总而的来说,本文报导了首款口服活性特异性JAK3抑制剂的设计。用化合物11阐述了如何在不增加化学反应性的情况下达到改善kinact的目的。通过哌啶环甲基取代的远程立体效应,实现了活性与降低GST介导的清除率相结合的优化目的。化合物11的高代谢稳定性以及它的整体特征都使得它适于口服。通过激酶组、HSA和人肝肝细胞谱分析证实化了合物11对其他含有半胱氨酸的蛋白质具有极低的反应性,这说明了特异性抑制JAK3的设计获得了成功。一系列体外和体内实验证实了化合物11对JAK3抑制的效果,解决了仅抑制JAK3(作为JAK1 / JAK3激酶异二聚体对中的伴侣)是否就足以抑制其通过γ-共链受体介导的细胞因子信号转导的争论。目前,化合物11已经进入到治疗炎症疾病的临床研究阶段。
文献
- Norman, P. Selective JAK1 inhibitor and selective TYK2 inhibitor patents. Expert Opin. Ther. Pat. 2012, 22, 1233−1249.
- Wilson, L. J. Recent patents in the discovery of small molecule inhibitors of JAK3. Expert Opin. Ther. Pat. 2010, 20, 609−623.
- Clark, J. D.; Flanagan, M. E.; Telliez, J. B. Discovery and development of Janus kinase (JAK) inhibitors for inflammatory diseases. J. Med. Chem. 2014, 57, 5023−5038.
- Thoma, G.; Drückes, P.; Zerwes, H. G. Selective inhibitors of the Janus kinase Jak3-Are they effective? Bioorg. Med. Chem. Lett. 2014, 24, 4617−4621.
- (7)Thorarensen, A.; Banker, M. E.; Fensome, A.; Telliez, J. B.; Juba, B.; Vincent, F.; Czerwinski, R. M.; Casimiro-Garcia, A. ATP-mediated kinome selectivity: the missing link in understanding the contribution of individual JAK Kinase isoforms to cellular signaling. ACS Chem. Biol. 2014, 9, 1552−1558.
- (8) Alicea-Vela̅zquez, N. L.; Boggon, T. J. The use of structural biology in Janus kinase targeted drug discovery. Curr. Drug Targets 2011, 12, 546−555.
- (9) Goedken, E. R.; Argiriadi, M. A.; Banach, D. L.; Fiamengo, B. A.; Foley, S. E.; Frank, K. E.; George, J. S.; Harris, C. M.; Hobson, A. D.; Ihle, D. C.; Marcotte, D.; Merta, P. J.; Michalak, M. E.; Murdock, S. E.; Tomlinson, M. J.; Voss, J. W. Tricyclic covalent inhibitors selectively target Jak3 through an active-site thiol. J. Biol. Chem. 2015, 290, 4573− 4589.
- (10) Tan, L.; Akahane, K.; McNally, R.; Reyskens, K. M.; Ficarro, S.B.; Liu, S.; Herter-Sprie, G. S.; Koyama, S.; Pattison, M. J.; Labella, K. M.; Johannessen, L.; Akbay, E. A.; Wong, K. K.; Frank, D. A.; Marto, J. A.; Look, A. T.; Arthur, S.; Eck, M. J.; Gray, N. S. Development of selective covalent JAK3 inhibitors. J. Med. Chem. 2015, 58, 6589− 6606.
- (20) Telliez, J. B.; Dowty, M. E.; Wang, L.; Jussif, J.; Lin, T.; Li, L.; Moy, E.; Balbo, P.; Li, W.; Zhao, Y.; Crouse, K.; Dickinson, C.; Symanowicz, P.; Hegen, M.; Banker, M. E.; Vincent, F.; Unwalla, R.;Liang, S.; Gilbert, A. M.; Brown, M. F.; Hayward, M.; Montgomery, J.; Yang, X.; Bauman, J.; Trujillo, J. I.; Casimiro-Garcia, A.; Vajdos, F. F.; Leung, L.; Geoghegan, K. F.; Quazi, A.; Xuan, D.; Jones, L. H.; Hett, E.; Wright, K.; Clark, J. D.; Thorarensen, A. Discovery of a novel JAK3-selective inhibitor: Functional differentiation of JAK3-selective inhibition over pan-JAK or JAK1-selective inhibition. ACS Chem. Biol. 2016, 11, 3442−3451.
- (11) Chrencik, J. E.; Patny, A.; Leung, I. K.; Korniski, B.; Emmons, T. L.; Hall, T.; Weinberg, R. A.; Gormley, J. A.; Williams, J. M.; Day, J. E.; Hirsch, J. L.; Kiefer, J. R.; Leone, J. W.; Fischer, H. D.; Sommers, C. D.; Huang, H. C.; Jacobsen, E. J.; Tenbrink, R. E.; Tomasselli, A. G.; Benson, T. E. Structural and thermodynamic characterization of the TYK2 and JAK3 kinase domains in complex with CP-690550 and CMP-6. J. Mol. Biol. 2010, 400, 413−433.
- (12) We evaluated the conformational preference of compounds 4 and 5 with high-level DFT calculations (ref) using the M06-2X method and 6-31G* basis set. These calculations suggest that conformer A where the N−H group is oriented toward the pyrrole ring is energetically favored over the alternative rotational isomer in the gas phase by ∼0.6 kcal/mol and resulting in a 70:30 distribution for the two conformers. In contrast for the N-Me group both conformers are almost equally populated in the gas phase with the conformational energy difference being only 0.02 kcal/mol.
- (13) Maurer, T. S.; Tabrizi-Fard, M. A.; Fung, H. L. Impact of mechanism-based enzyme inactivation on inhibitor potency: implications for rational drug discovery. J. Pharm. Sci. 2000, 89, 1404−1414.
- (14) Thorarensen, A. Unpublished results.
- (15) Flanagan, M. E.; Abramite, J. A.; Anderson, D. P.; Aulabaugh, A.; Dahal, U. P.; Gilbert, A. M.; Li, C.; Montgomery, J.; Oppenheimer, S. R.; Ryder, T.; Schuff, B. P.; Uccello, D. P.; Walker, G. S.; Wu, Y.; Brown, M. F.; Chen, J. M.; Hayward, M. M.; Noe, M. C.; Obach, R. S.; Philippe, L.; Shanmugasundaram, V.; Shapiro, M. J.; Starr, J.; Stroh, J.; Che, Y. Chemical and computational methods for the characterization of covalent reactive groups for the prospective design of irreversible inhibitors. J. Med. Chem. 2014, 57, 10072−10079.
- (18) Schwartz, P. A.; Kuzmic, P.; Solowiej, J.; Bergqvist, S.; Bolanos, B.; Almaden, C.; Nagata, A.; Ryan, K.; Feng, J.; Dalvie, D.; Kath, J. C.; Xu, M.; Wani, R.; Murray, B. W. Covalent EGFR inhibitor analysis reveals importance of reversible interactions to potency and mechanisms of drug resistance. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 173−178.
- (19) Siewert, E.; Müller-Esterl, W.; Starr, R.; Heinrich, P. C.; Schaper, F. Different protein turnover of interleukin-6-type cytokine signaling components. Eur. J. Biochem. 1999, 265, 251−257.
- (20) Telliez, J. B.; Dowty, M. E.; Wang, L.; Jussif, J.; Lin, T.; Li, L.; Moy, E.; Balbo, P.; Li, W.; Zhao, Y.; Crouse, K.; Dickinson, C.; Symanowicz, P.; Hegen, M.; Banker, M. E.; Vincent, F.; Unwalla, R.;Liang, S.; Gilbert, A. M.; Brown, M. F.; Hayward, M.; Montgomery, J.; Yang, X.; Bauman, J.; Trujillo, J. I.; Casimiro-Garcia, A.; Vajdos, F. F.; Leung, L.; Geoghegan, K. F.; Quazi, A.; Xuan, D.; Jones, L. H.; Hett, E.; Wright, K.; Clark, J. D.; Thorarensen, A. Discovery of a novel JAK3-selective inhibitor: Functional differentiation of JAK3-selective inhibition over pan-JAK or JAK1-selective inhibition. ACS Chem. Biol. 2016, 11, 3442−3451.
- (21) Meyers, M. J.; Long, S. A.; Pelc, M. J.; Wang, J. L.; Bowen, S. J.; Schweitzer, B. A.; Wilcox, M. V.; McDonald, J.; Smith, S. E.; Foltin, S.; Rumsey, J.; Yang, Y. S.; Walker, M. C.; Kamtekar, S.; Beidler, D.; Thorarensen, A. Discovery of novel spirocyclic inhibitors of fatty acid amide hydrolase (FAAH). Part 2. Discovery of 7-azaspiro[3.5]nonane urea PF-04862853, an orally efficacious inhibitor of fatty acid amide hydrolase (FAAH) for pain. Bioorg. Med. Chem. Lett. 2011, 21, 6545− 6553.
- (22) Kim, K. H.; Maderna, A.; Schnute, M. E.; Hegen, M.; Mohan, S.; Miyashiro, J.; Lin, L.; Li, E.; Keegan, S.; Lussier, J.; Wrocklage, C.; Nickerson-Nutter, C. L.; Wittwer, A. J.; Soutter, H.; Caspers, N.; Han, S.; Kurumbail, R.; Dunussi-Joannopoulos, K.; Douhan, J., 3rd; Wissner, A. Imidazo[1,5-a]quinoxalines as irreversible BTK inhibitors for the treatment of rheumatoid arthritis. Bioorg. Med. Chem. Lett. 2011, 21, 6258−6263.
- (23) Shibata, Y.; Chiba, M. The role of extrahepatic metabolism in the pharmacokinetics of the targeted covalent inhibitors afatinib, ibrutinib, and neratinib. Drug Metab. Dispos. 2015, 43, 375−384.
- (24) Leung, L.; Yang, X.; Strelevitz, T. J.; Montgomery, J.; Brown, M. F.; Zientek, M. A.; Banfield, C.; Gilbert, A. M.; Thorarensen, A.; Martin, E.; Dowty, M. E. Clearance prediction of targeted covalent inhibitors by in vitro-in vivo extrapolation of hepatic and extrahepatic clearance mechanisms. Drug Metab. Dispos. 2017, 45,1−7.
- (25) Carron, C. P.; Trujillo, J. I.; Olson, K. L.; Huang, W.; Hamper, B. C.; Dice, T.; Neal, B. E.; Pelc, M. J.; Day, J. E.; Rohrer, D. C.; Kiefer, J. R.; Moon, J. B.; Schweitzer, B. A.; Blake, T. D.; Turner, S. R.; Woerndle, R.; Case, B. L.; Bono, C. P.; Dilworth, V. M.; FunckesShippy, C. L.; Hood, B. L.; Jerome, G. M.; Kornmeier, C. M.; Radabaugh, M. R.; Williams, M. L.; Davies, M. S.; Wegner, C. D.; Welsch, D. J.; Abraham, W. M.; Warren, C. J.; Dowty, M. E.; Hua, F.; Zutshi, A.; Yang, J. Z.; Thorarensen, A. Discovery of an oral potent selective inhibitor of hematopoietic prostaglandin D synthase (HPGDS). ACS Med. Chem. Lett. 2010, 1, 59−63.
- (26) Casimiro-Garcia, A. Unpublished results.
- (27) Compound 11 is commercially available via Sigma Aldrich (catalog no. PZ0316).
- (28) Hett, E. Unpublished results.
- (29) Liu, Q.; Sabnis, Y.; Zhao, Z.; Zhang, T.; Buhrlage, S. J.; Jones, L. H.; Gray, N. S. Developing irreversible inhibitors of the protein kinase cysteinome. Chem. Biol. 2013, 20, 146−159.



{kind=link}