
摘要:本文以JAK3共价抑制剂PF-06651600为例,演示了如何用Ligandscout的药效团片段筛选方法对片段数据库进行虚拟筛选,来识别潜在激酶铰链结合片段;还演示如何进一步用AutoT&T自动将打分更优的片段裁剪、移植到先导化合物上,从而实现JAK3共价抑制剂铰链结合片段的跃迁。
1. 前言
之前我们以辉瑞公司的选择性JAK3共价抑制剂PF-06651600(图1)为例,演示了如何用基于反应的组合库设计策略进行保留铰链区结合片段(图1橘黄色高亮部分)与丙烯酰Michael受体结构片段(图1红色高亮部分)而引入不同的连接臂(图1黄色高亮部分)实现多样的潜在共价抑制剂组合库构建[1]。我们还进一步演示了如何用LigandScout药效团技术与OpenEye的FRED分子对接技术对枚举的组合库进行虚拟筛选,并证明药效团与分子对接方法不仅可以将活性与非活性的化合物区分开,而且还能发现新的、令人感兴趣的结构[2]。
图1. 辉瑞公司的选择性JAK3共价抑制剂PF-06651600的化学结构
LigandScout基于药效团片段的虚拟筛选策略也适合于对共价抑制的弹头部分进行虚拟筛选。比如在Schulz等人[3]的研究中,采用LigandScout基于结构的方法识别了肠病毒3 CLPro共价键结合的3D药效团模型;并用包含33个活性化合物、1771个decoy化合物的数据集对该药效团模型进行验证,发现该模型具有非常好的敏感性与专一性;对一个3000个化合物的数据库进行虚拟筛选发现了靶向肠病毒3CLPro Cys的共价结合的不可逆抑制剂;最后通过骨架跃迁、结构优化发现了化学稳定的共价抑制剂。
而针对激酶铰链区结合片段的筛选,已经有很多的实验筛选与计算筛选的文献报道。比如Noji等人[4]在JAK3抑制剂Delgocitinib(开发代号JTE-052)的发现阶段,就从现有已知的铰链结合片段里系统性地进行实验筛选适于JAK3的铰链结合片段。
关于铰链结合片段的计算识别有两个目的:现有激酶铰链片段再利用(repurpose)与发现全新的铰链结合片段。已经有许多计算方法识别铰链结合片段的文献报道,在Rachman等人[5]的文章中有详细的综述。比如,黄牛课题组的Yanli Wang等人[6]以p38α MAPK作为模型体系,详细报道了如何用基于结构的虚拟筛选方法识别全新的铰链区结合剂。在Yanli Wang等人的研究中,首先用分子对接方法虚拟筛选一个含107238化合物的内部数据库;然后用两个专用的激酶结构过滤器对命中化合物进行过滤,以出去不满足条件的化合物;然后用基于能量最小化的MM-GB/SA对化合物打分,再用激酶结构过滤器进行一轮的过滤,保留500个打分最优的化合物进入下一轮计算评估;最后用基于分子动力学模拟的MM-GB/SA进一步对化合物进行打分,最终选择了8个化合物进入最实验测试。Rachman等人[5]采用一个相对比较简洁的方法,首先用药效团约束的分子对接方法进行虚拟筛选,以确保化合物与铰链区有特定的氢键相互作用;然后再用DUck(Dynamic undocking)方法评估这些氢键断裂的阻力以优选片段。经过实验验证,Rachman等人发现了一个全新的hinge结合片段8-amino-2H-isoquinolin-1-one(MR1)。STD-NMR实验表明,MR1可以与MELK的铰链区直接结合,有望用于之后的分子生长以设计新APT竞争性抑制剂。
如前所述,LigandScout基于药效团的片段筛选可以快速、高效地对片段库进行虚拟筛选,而AutoT&T又善于将裁剪与移植,我非常急切地想知道两者的组合是否可以帮助我们实现激酶抑制剂的铰链结合片段的发现、建立集中库并最终用于新的化合物的发现。因此本文的目的之一是演示如何用Ligandscout的药效团片段筛选方法来实现上面的激酶铰链结合片段的虚拟筛选;本文另一个目的是演示如何进一步用AutoT&T自动识别打分更优的铰链区结合片段,并将之裁剪、移植到先导化合物上,从而建立激酶抑制剂集中库、实现铰链结合片段跃迁。具体的讲,本文以JAK3共价抑制剂PF-06651600的铰链结合片段进行跃迁为例,演示如何实现上述的铰链区结合片段的虚拟筛选、裁剪与移植。
2. 计算实验操作过程
2.1 药效团模型的建立
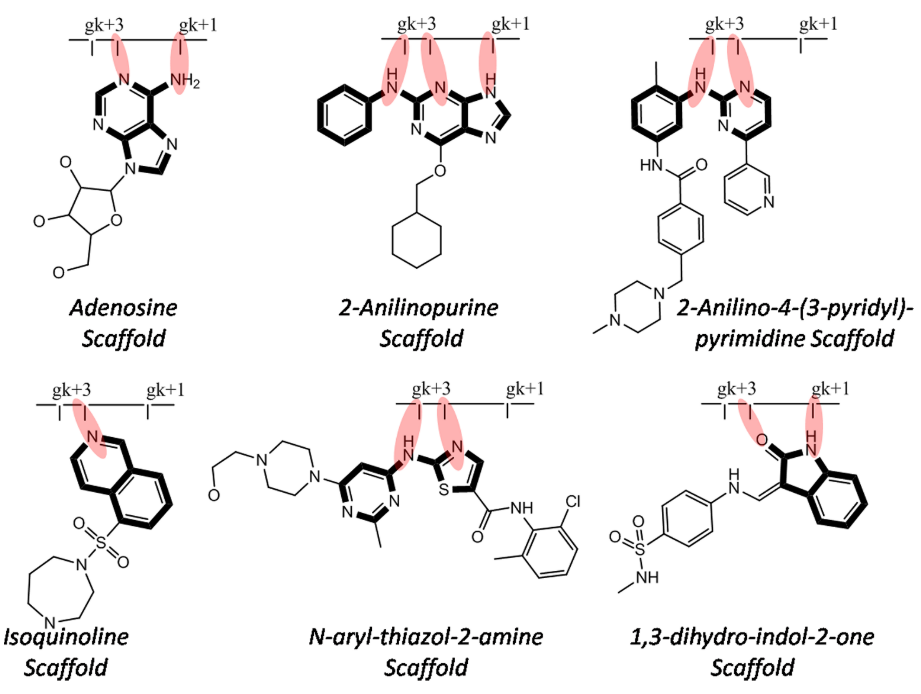
图2. Yanli Wang等人[6]总结的激酶结合剂代表性的结合模式
根据Yanli Wang等人[6]总结的激酶铰链区结合剂代表性的结合模式(图2),如果仅用一个药效团模型无法覆盖所有的结合模式。为了方便演示而又尽可能覆盖更多的结合模式,我们用源自PDB 5toz与4z16识别的药效团为例来设计尽可能覆盖更多结合模式的query进行虚拟筛选。
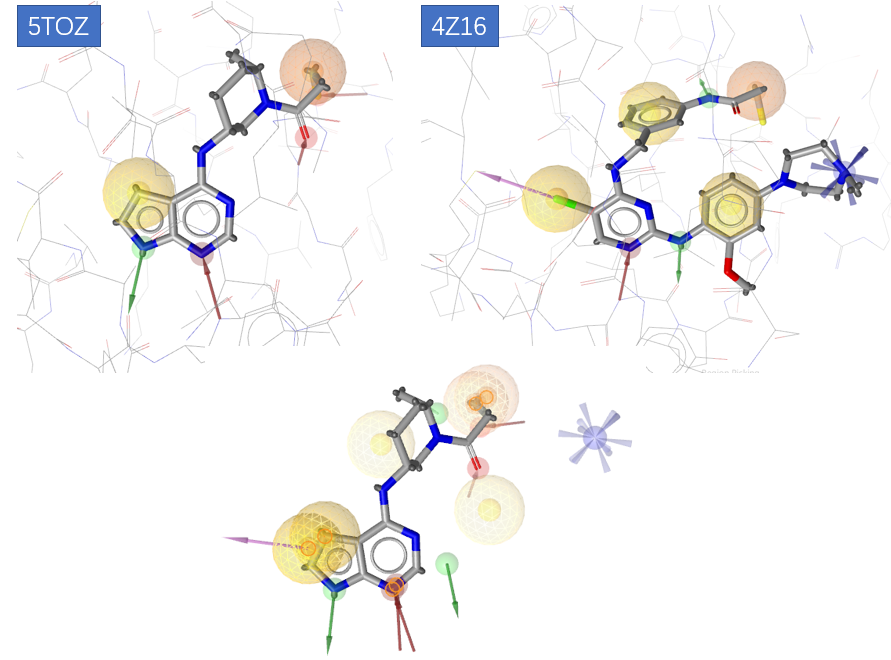
图3. 从PDB 5toz与4z16识别的基于结构的药效团模型。上左:PDB 5TOZ;上右:PDB 4Z16;下:两者的叠合(其中非铰链区部分做了虚化处理)。
首先,用ligandscout基于结构的方法从PDB 5toz与4z16识别药效团(不考虑与水的相互作用),结果见图3(上)。将两种药效叠合(见图3下),可以发现,在铰链区这两个药效团模型具有公共的药效团特征:一个氢键受体(红色)、一个疏水中心(黄色球。而在公共的氢键受体两侧,各自有一个特有的氢键供体(绿色)。
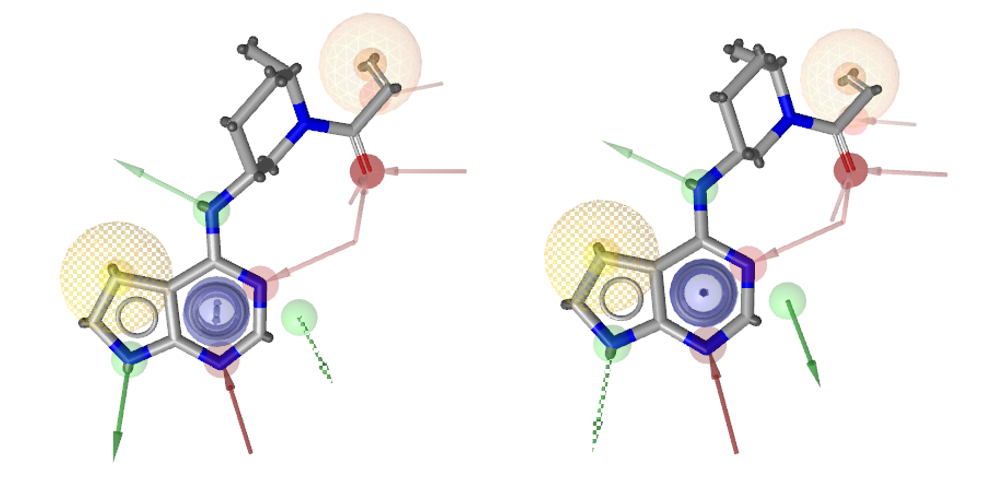
图4. 用于虚拟筛选的药效团模型
最终设计了两个代表性药效团模型用于铰链区结合剂的虚拟筛选,如图4所示,该模型包含了一个氢键受体、一个供体、一个芳香环、一个可选的供体与疏水中心。在虚拟筛选时,这两个药效模型以boolean操作符号or组合进行虚拟筛选(图5),以确保覆盖更多可能的已知结合模式。
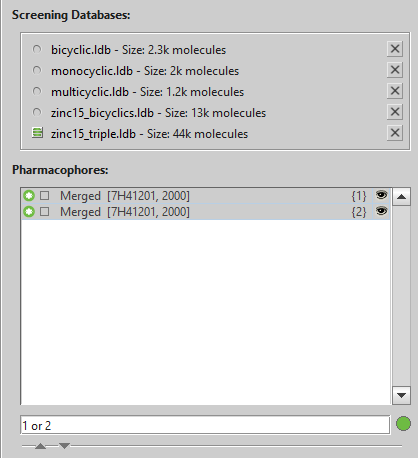
图5. 用boolean操作符号“or”将两种药效团组合进行虚拟筛选
上述将两个药效团模型进行merge操作,意在演示,还有其它简洁的方法可以得到同样的效果。比如,将一个另一个配体插入(docking)到同一个结合口袋,再生成药效团模型;或者选择一个合适的配体-蛋白复合物结构生成药效团模型。
2.2 数据库的准备
数据库取自Zhao Hongtao等人[7,8]从ChEMBL采集的激酶铰链结合片段,其包含10,302个不重复的含有至少一个环的铰链结合片段,分成三个子库:单环、双环与三环。用LigandScout的数据库准备工具以icon best模式进行构象搜索。
Zhao Hongtao等人[7,8]碎片化方法尤其适合于激酶抑制剂的铰链结合片段,建议在真正的研究中用该方法重新对感兴趣的化合物库进行碎片化,然后再进行虚拟筛选。本文仅是演示的目的,因此就已经准备好的片段库。
2.3 虚拟筛选
如图5所示,在LigandScout/Virtual screening里通过boolean操作符“or”使用2.1述所的两个药效团模型对2.2准备的数据库进行虚拟筛选(使用get the best matching conformation模式),结果命中1205个片段。
对命中的片段进一步用Binding Affinity score打分,仅保留打分值(Binding affinity score)小于-15kJ/mol的化合物,总共得到891个片段。LigandScout的Binding affinity score是Hyde score的一种实现,根据Reulecke等人(2008)研究[8],对于片段binder来说,打分值优于-15~-25kJ/mol的片段具有更高的概率为真binder。
2.4 新化合物生成:自动裁剪与移植
在本次的裁剪、移植连接过程中,将被替换基团定义为图1橘黄色部分。因此将连接橘黄色与黄色区域的C16-N3单键定义为优化位点。虽然这样会错过一些其它优化位点的替换方式,但是可以简化结果。LinkLeadOpt是AutoT&T实现片段裁剪、连接策略的工具,命令如下:
1 2 3 4 5 6 | LinkLeadOpt -l 5toz_ligand.sdf \ -vs hinge-fragment-hits.sdf \ -p 5toz_C908G_DRY.pdb \ -out jak3_linked_LIBO.sdf \ -c '(3,16) \ -ih |
其中,参数-l读入先导化合物(本例中为PDB 5TOZ的共晶配体: 5toz_ligand.sdf); 参数-vs读入参考分子库,也就是在2.2节命中的片段(hinge-fragment-hits.sdf); 参数-p读入共晶结构的蛋白(PDB 5toz,为了避免配体与Cys残基发生碰撞而影响docking打分,进行了C809G的突变操作);参数-c指定了优化位点,若剪切下来的片段在打分上优于第3与16序号原子(即为图1橘黄色与黄色区域C16-N3键的原子序号)所在的片段,则进行片段替换;参数-out用来指定输出文件的名称;-ih表示C-H参与替换的键匹配。
结果LinkLeadOpt生成了226个新的化合物,其铰链结合片段源自现有激酶铰链结合片段。用AutoT&T的Cluster命令进行聚类分析:
1 2 3 4 | /public/apps/att2/bin-linux/Cluster \ -i jak3_linked_LIBO.sdf \ -o jak3_cluster.log \ -ap -sca 0.5 |
其中-ap参数用来指示cluster命令采用原子对相似性法进行聚类、-sca指示聚类分析的距离截断值为0.5。
结果表明,生成的226个化合物归属于20个不同结构骨架类别,从化学结构上看,连接策略确实得到了结构多样的化合物。
在新生成的化合物中,包含了先导化合物PF-06651600本身及其甲基化衍生物(见图6左),以及与先导化合物非常相似的化合物(如图6右),它们与靶标(去水的“干”蛋白“)的相互作用模式上也一样。
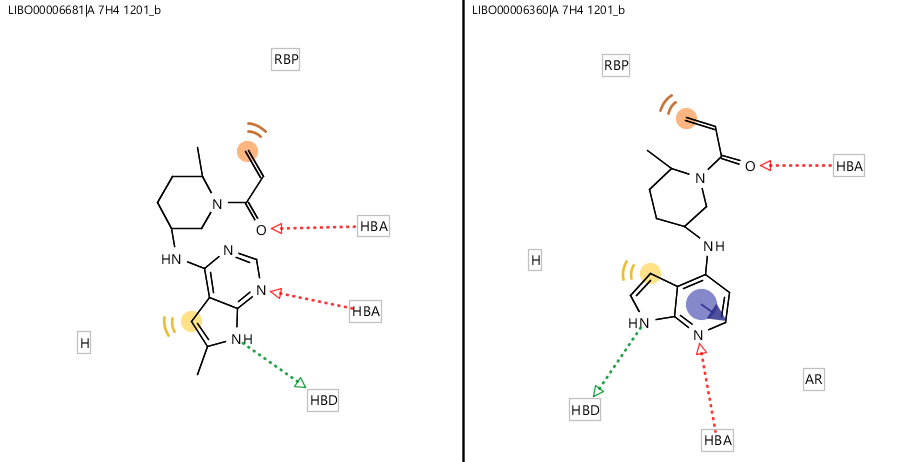
图6. 分子连接生成的化合物获得了与先导化合物PF-06651600的衍生物(左)以及类似物(右),它们与靶标的相互作用模式是一样的。
我们还可以发现,如图7所示,还生成了与铰链区残基发生3次氢键相互作用的化合物,它们与铰链区的相互作用模式不同于先导化合物,这也说明预计的筛选策略起到了效果。
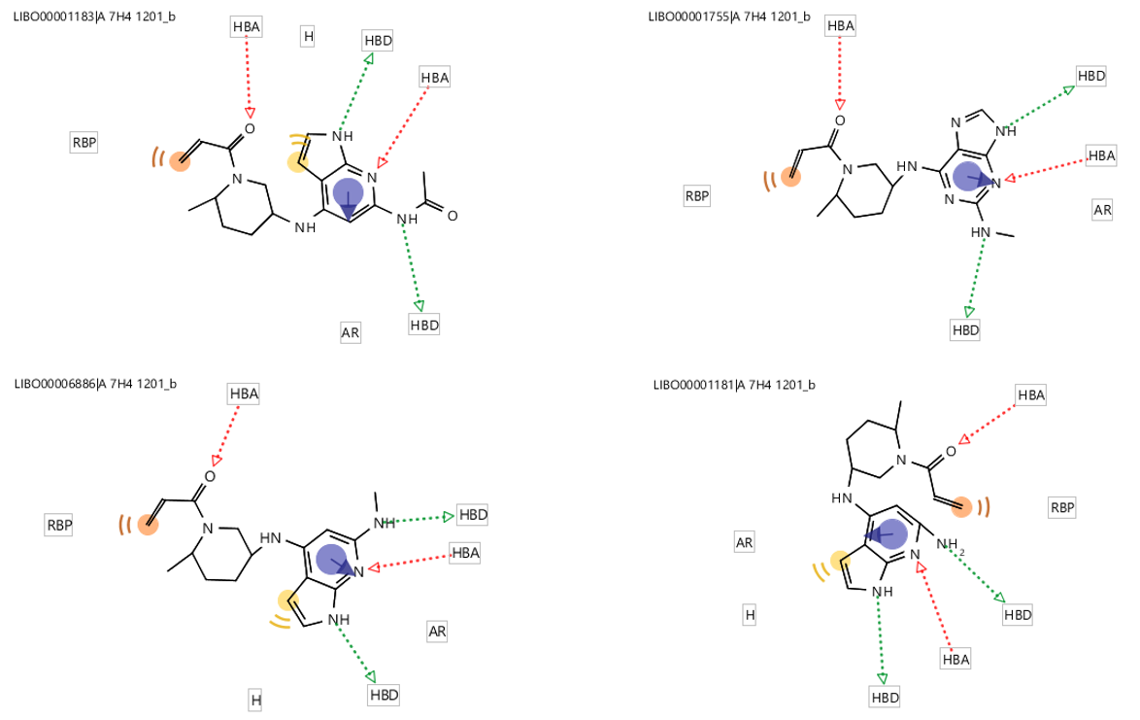
图7. 4个铰链区结合片段结构与铰链区残基发生三次氢相互作用的化合物
此外,还命中了区别于先导化合物与靶标相互作用模式不同的另一个药效团模型(图4右)的化合物,如图8所示。
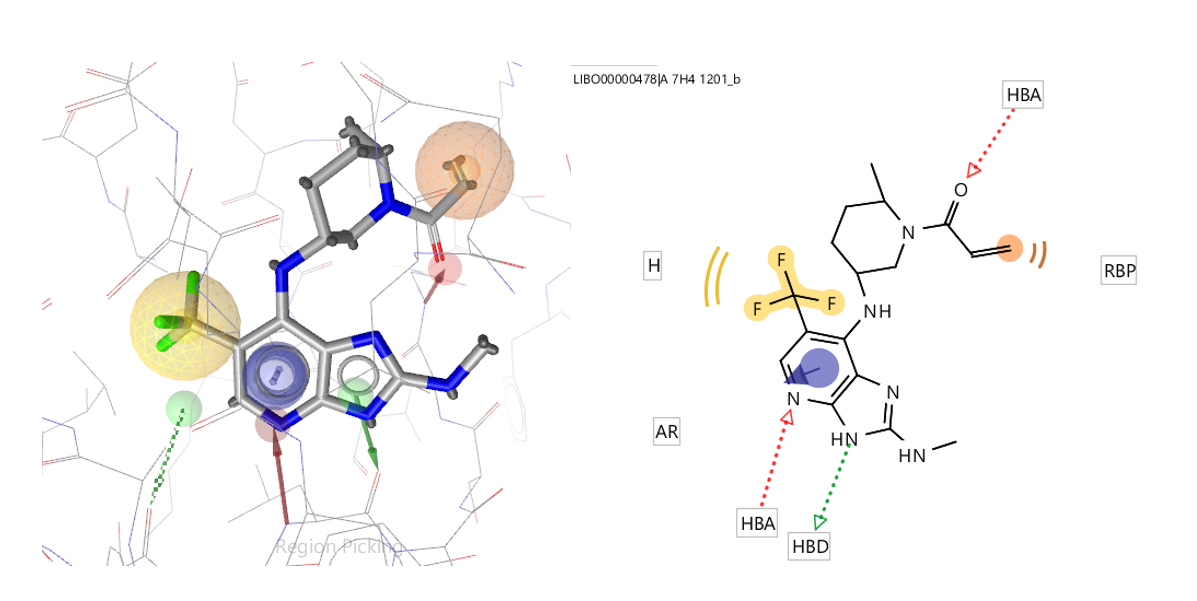
图8. 一个不同于先导化合物结合模式的的化合物示例
这说明,我们的虚拟筛选与连接策略生成的新化合物在相互作用模式上,既包含了与先导化合物一致的化合物,也包含了新颖相互作用模式的化合物。
为了确保生成的结构在能量上是合理的,需要在蛋白结合位点里对化合物结构进行力场优化,可以用Ligandscout的Molecule Minization来进行此类操作。优化之后,化合物的结构有可能偏离初始的位置,甚至不能与药效团匹配,因此有必要进行再次打分以确保化合物满足预设的结合模式。
3. 小结
总的来说,本次针对JAK3先导化合物PF-06651600进行的铰链结合区的结合片段的药效团虚拟筛选,总共从现有激酶铰链结合片段里发现了1205个片段;并进一步被Binding Affinity打分优选891个片段,这些片段有更高的概率可能为真binder。
用AutoT&T的连接策略对PF-06651600铰链结合片段进行的基团替换实验生成了226个新结构,聚类分析表明其具有结构多样性,覆盖了20个结构类别。从结合模式上看,生成的化合物与铰链区的相互作用模式包含了预先设定的3种模式,其中两种有别于先导化合物PF-06651600,这表明我们的方法可以发现具有新作用模式的化合物。
Zhao Hongtao等人[7,8]碎片化方法尤其适合于激酶抑制剂的铰链结合片段,建议在真正的研究中用该方法对感兴趣的化合物库进行碎片化,然后再进行虚拟筛选。
4. 文献
- 肖高铿(2018-07-05).墨灵格的博客. KNIME教程|基于反应的组合库生成
- 肖高铿(2018-03-22).墨灵格的博客. 用常规分子对接进行共价抑制剂的虚拟筛选
- Schulz, R., Atef, A., et al.Phenylthiomethyl Ketone-Based Fragments Show Selective and Irreversible Inhibition of Enteroviral 3C Proteases. Journal of Medicinal Chemistry,2018, 61(3):1218–1230. https://doi.org/10.1021/acs.jmedchem.7b01440
- Noji, S.; Hara, Y.; Miura, T.; Yamanaka, H.; Maeda, K.; Hori, A.; Yamamoto, H.; Obika, S.; Inoue, M.; Hase, Y.; et al. Discovery of a Janus Kinase Inhibitor Bearing a Highly Three-Dimensional Spiro Scaffold: JTE-052 (Delgocitinib) as a New Dermatological Agent to Treat Inflammatory Skin Disorders. J. Med. Chem. 2020, 63 (13), 7163–7185. https://doi.org/10.1021/acs.jmedchem.0c00450.
- Rachman, M.; Bajusz, D.; Hetényi, A.; Scarpino, A.; Mero, B.; Egyed, A.; Buday, L.; Barril, X.; Keseru, G. M. Discovery of a Novel Kinase Hinge Binder Fragment by Dynamic Undocking. RSC Med. Chem. 2020, 11 (5), 552–558. https://doi.org/10.1039/c9md00519f.
- Wang, Y.; Sun, Y.; Cao, R.; Liu, D.; Xie, Y.; Li, L.; Qi, X.; Huang, N. In Silico Identification of a Novel Hinge-Binding Scaffold for Kinase Inhibitor Discovery. J. Med. Chem. 2017, 60 (20), 8552–8564. https://doi.org/10.1021/acs.jmedchem.7b01075.
- Zhao, H.; Caflisch, A. Current Kinase Inhibitors Cover a Tiny Fraction of Fragment Space. Bioorganic Med. Chem. Lett. 2015, 25 (11), 2372–2376. https://doi.org/10.1016/j.bmcl.2015.04.005.
- Kinase Hinge-Binding Fragments. Retreived from http://www.lephar.com/download.htm
- Reulecke, I.; Lange, G.; Albrecht, J.; Klein, R.; Rarey, M. Towards an Integrated Description of Hydrogen Bonding and Dehydration: Decreasing False Positives in Virtual Screening with the HYDE Scoring Function. ChemMedChem 2008, 3 (6), 885–897.


