
摘要:本文描述了如何联合使用LigandScout与REINVENT实现深度生成模型 (DGM) 设计具有所需药效团的DDR1抑制剂。 DDR1 是一种由基质胶原激活的受体酪氨酸激酶,与癌症、纤维化和缺氧等疾病有关。本文描述了使用DGM生成化合物、然后进行合成、并测试其抑制活性。发现化合物 3、4 和 7a 具有亚微摩尔抑制活性。其中最有效的化合物3的IC50为92.5nM。此外,预计这些化合物会与DDR1相互作用,具有源自已知DDR1抑制剂所需的药效团。合成和实验测试结果表明,我们的从头设计策略用于苗头化合物的识别以及骨架跃迁是可行的。
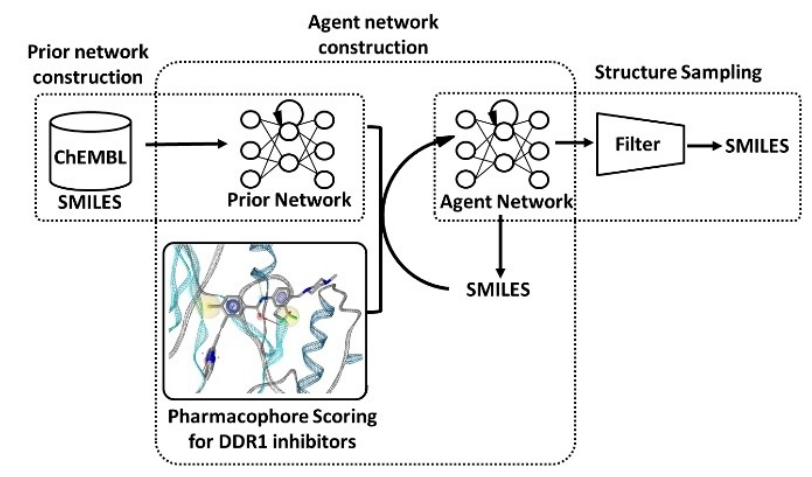
图1. DDR1抑制剂识别的工作流
原文:Yoshimori, A.; Asawa, Y.; Kawasaki, E.; Tasaka, T.; Matsuda, S.; Sekikawa, T.; Tanabe, S.; Neya, M.; Natsugari, H.; Kanai, C. Design and Synthesis of DDR1 Inhibitors with a Desired Pharmacophore Using Deep Generative Models. ChemMedChem 2021, 16 (6), 955–958. https://doi.org/10.1002/cmdc.202000786.
编译:肖高铿
深度生成模型(Deep generative models,DGM))已成功应用于图像生成[1]、 语言翻译[2]以及其它领域[3]。近年来,在从头药物设计中使用DGM生成化学结构受到了越来越多的关注[4]。几个小组报告了使用DGM进行苗头化合物识别的成功例子[5,6]。尽管DGM能够生成具有所需性质的分子,但大多数性质并不包含诸如形状和药效团之类的3D信息[4]。众所周知,源自3D形状和/或药效团的性质在药物设计过程中非常有用[7]。因此,我们构建了用于生成具有所需药效团分子的DGM。DGM构建的过程已在别处发表有论文描述[8]。简而言之,该方法由三个步骤组成(如图1所示),包括:先验网络构建(prior network construction)、代理网络构建(agent network construction)和结构采样(structure sampling)。首先,用ChEMBL[10]的SMILES字符串[9]来训练先验网络。训练后,先验网络具有生成有效SMILES字符串的能力。接下来,使用强化学习(reinforcement learning)来训练代理网络。训练后,药效团打分的概率分布从先验网络的概率分布向期望的药效团打分分布迁移。在第三步中,经过训练的代理网络生成可能具有所需药效团的SMILES字符串。其中,使用REINVENT[11]实现先验和代理网络的构建,使用LigandScout 4.4[12]生成药效团并计算药效团打分。
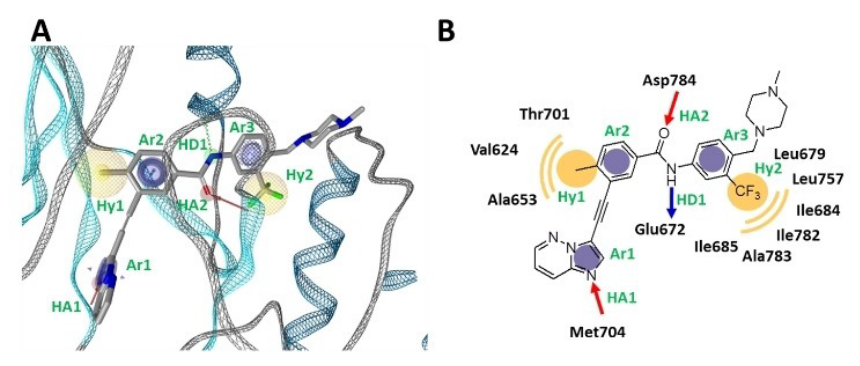
图2. (A)DDR1抑制剂的药效团模型;(B)DDR1抑制剂药效团模型的2D示意图
DDR1(Discoidin domain receptor 1)是一种胶原蛋白激活受体酪氨酸激酶,是癌症[13]、纤维化[14]和缺氧[15]等多种人类疾病的潜在治疗靶点。
Ponatinib与DDR1激酶结构域的共晶复合结构PDB 3ZOS用来构建所需的DDR1药效团(图 2A)[16]。 在这里,确定了抑制剂的八个药效团特征(图 2B):三个芳香环(Ar1、Ar2、Ar3)、两个疏水中心(Hy1、Hy2)、两个氢键受体(HA1、HA2)和一个氢供体(HD1)特征。此外,还使用了从晶体结构获得的排除体积球。药效团打分函数设置为“Relative Pharmacophore-Fit”;最大的被忽略特征数设置为“1”,并且Hy1、Hy2、Ar3和HD1特征设置为“可选特征(optional feature)”。相对药效团拟合(Relative Pharmacophore-Fit,rel.SFCR)如方程 (1) 定义[8,12]:
[latexpage]
\begin{equation} \label{eq:poly}
rel.S_{FCR} = \frac{S_{FCR}}{10 \times (N_{MFP}+1)}
\end{equation}
\[
S_{FCR} = 10 \times N_{MFP} + S_{RMS}
\]
\[
S_{RMS} = 9 – 3 \times Min(RMS_{FP},3)
\]
其中SFCR是特征计数/RMS距离分数;NMFP是在几何上匹配的特征对数量;SRMS是匹配特征对的RMS距离分数,取值范围为[0,9];RMSFP是匹配特征对距离的RMS。LigandScout 4.4的idbgen[17]以“iCon Fast”模式用于代理网络生成结构的构象生成。
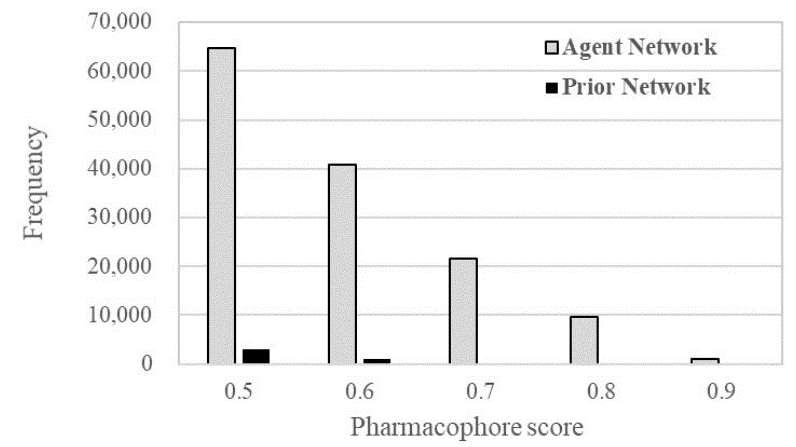
图3. 先验网络与代理网络生成结构的药效团打分值分布。横轴药效团打分值小于0.5的部分被忽略掉,因为所有命中化合物的药效团打分都大于0.5
代理网络用所需的药效团和先验网络进行训练。代理网络的训练使用64的批量大小(Batch Size)、Adam优化器完成10,000 步的训练。其他参数使用REINVENT中的默认值。训练结束后,进行结构采样。在640,000个SMILES字符串的结构采样期间生成了570,542个有效的SMILES。以同样的方式,先验网络生成了588,240个有效的SMILES。生成的结构用LigandScout 4.4计算药效团打分。药效团打分的分布显示为图3中的直方图。在代理网络生成的有效SMILES字符串中有137,790个化合物的药效团打分大于0.5。在先验网络生成的有效SMILES字符串中,只有4,306个化合物的药效团打分值高于0.5。这一结果表明,代理网络能以较高的频率产生满足DDR1抑制剂药效团的结构。
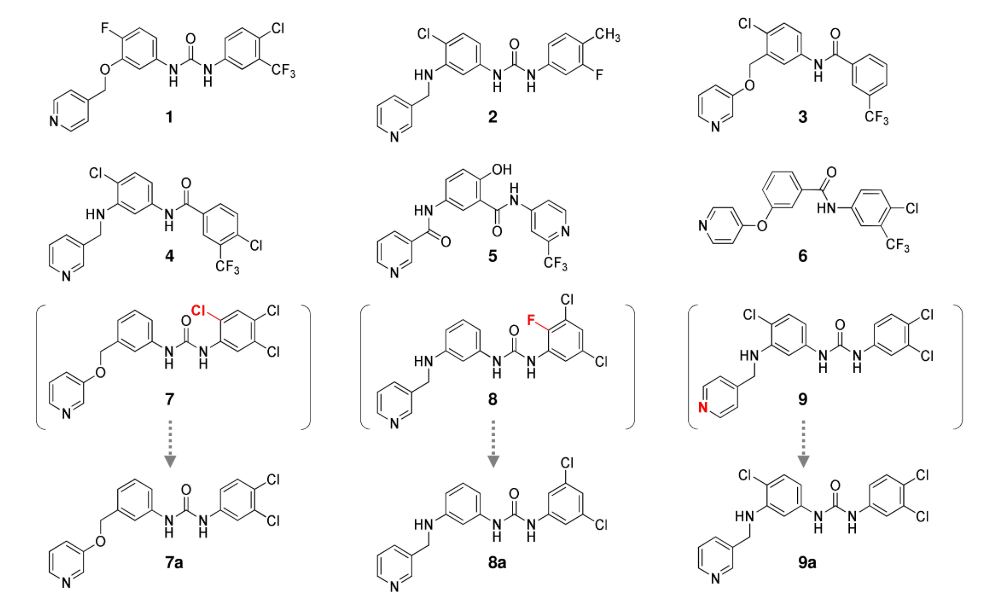
图4. 合成并进行DDR1抑制活性测试的化合物
为了对代理网络生成的结构进行过滤,将药效团打分值高于0.8的结构(10,694个结构)过滤出来,并用LigandScout 4.4中实现的iaffinity模块计算结合亲和力打分,将结合亲和力打分小于-37kJ/mol的化合物(4731个化合物)进一步挑选出来。对选定的化合物进行目视检查以确定要合成哪些化合物,同时考虑它们的药效团打分、结合亲和力打分和合成可行性。因此,选择了9种化合物 (1-9),如图 4 所示。在目视检查期间,对两种化合物 (7, 8) 进行了去除卤素原子的修饰,因为卤素对与DDR1的结合相互作用没有太大贡献。将化合物9的吡啶基氮位置从对位变为间位以与DDR1的铰链区形成氢键。最后,合成了9种化合物(1-6 和 7a、8a、9a)。9种化合物的合成总结在路线1-9中(见支持信息)。
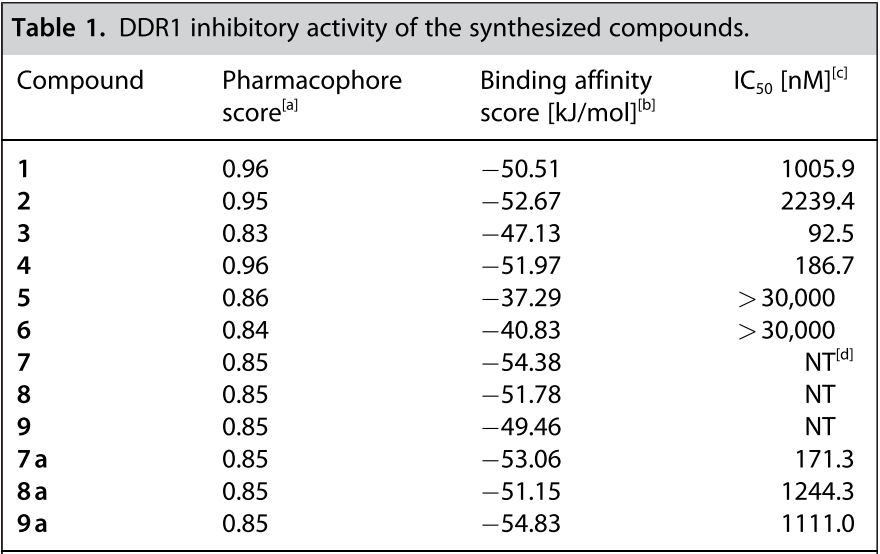
a.LigandScout 4.4计算的药效团打分值;b. LigandScout 4.4/iaffinity模块计算的结合亲和力; c.从半对数剂量-响应曲线确定50%抑制所需的化合物浓度(IC50),结果表示重复样品的平均值; d. NT=not tested。
对合成的化合物进行了DDR1抑制活性评价。激酶的活性测试采用芯片外迁移率变动分析法(Off-chip Mobility Shift Assay)进行测定(Carna Biosciences,Inc.,Kobe,Japan提供服务)(见支持信息),结果总结在表1中。在所测试的化合物中,化合物3对DDR1表现出令人感兴趣的两位数纳摩尔抑制活性(IC50=92.5nM)。图5A展示了化合物3与药效团匹配性以及相互作用模式,尽管存在轻微未对准的特征,但化合物3满足DDR1抑制剂的所有药效团特征。
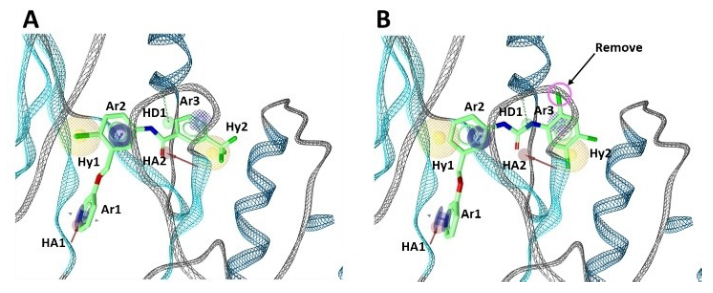
图5. 化合物3(A)、7(B)与药效团的匹配性及与DDR1的相互作用模式
还发现化合物4和7a具有有效的抑制活性(IC50值分别为186.7和171.3nM)。如图5B所示,化合物7a是将化合物7中的1,2,4-三氯苯片段的Cl原子去除设计而得。发现化合物1、2、8a、9a具有中等程度的DDR1抑制活性(IC50值分别为1005.9、2239.4、1244.3和1111.0nM)。化合物5和6根本不显示任何抑制活性。这两化合物的结合亲和力打分较高(-37.29和-40.83kJ/mol),表明结合亲和力较低。这些结果表明我们使用DGMS的策略有效地设计了DDR1抑制剂。
为了检查所生成的结构(化合物 1-6、7a-9a)是否已经在某些数据库中登记过,在 ChEMBL[10]和PubChem[18]中进行了结构搜索。我们发现化合物3在PubChem中登记过,其CID为58614959,并被注释为Raf激酶与p38 MAP激酶的抑制剂。
总的来说,我们能够使用DGMS设计具有所需药效基团的DDR1抑制剂。化合物3表现出有效的抑制活性,对DDR1的IC50值为92.5nM。通常,为了预测DGMS生成化合物的抑制活性,需要大量的实验抑制数据来构建准确的预测模型。然而,我们的策略只需要药效团信息来设计针对靶蛋白的抑制剂。因此,我们的策略可以用于药物发现过程的早期阶段。Ponatinib是用于治疗慢性髓系白血病的药物,其抑制DDR1的Kd值为1.3nM[16]。在本研究中,我们的药效团来源于与Ponatinib-DDR1激酶结构域共晶结构。合成化合物的骨架(图4)与Ponatinib的骨架不同(图2)。 因此,我们的策略也可以用于骨架跃迁。
为了确定合成哪些化合物,重要的是对代理网络生成的结构进行高效地过滤。我们现在正试图构建更实用的过滤方法,包括类药性打分[19]、ADMET性质[20]以及合成可行性打分[21]等标准。我们相信这种基于药效团的DGM策略可以应用于未来的各种药物发现项目。
参考文献
- H. Huang, P. S. Yu, C. Wang, arXiv:1803.04469v2 2018.
- M. Johnson, M. Schuster, Q. V. Le, M. Krikun, Y. Wu, Z. Chen, N. Thorat, F. Viégas, M. Wattenberg, G. Corrado, M. Hughes, J. Dean, Trans. Assoc. Comput. Linguist. 2017, 5, 339–351.
- R. de Bem, A. Ghosh, T. Ajanthan, O. Miksik, A. Boukhayma, N. Siddharth, P. Torr, Int. J. Comput. Vis. 2020, 128, 1537–1563.
- D. C. Elton, Z. Boukouvalas, M. D. Fugea, P. W. Chung, Mol. Syst. Des. Eng. 2019, 4, 828–849.
- D. Merk, L. Friedrich, F. Grisoni, G. Schneider, Mol. Inf. 2018, 37, 1700153.
- A. Zhavoronkov, Y. A. Ivanenkov, A. Aliper, M. S. Veselov, V. A. Aladinskiy, A. V. Aladinskaya, V. A. Terentiev, D. A. Polykovskiy, M. D. Kuznetsov, A. Asadulaev, Y. Volkov, A. Zholus, R. R. Shayakhmetov, A. Zhebrak, L. I. Minaeva, B. A. Zagribelnyy, L. H. Lee, R. Soll, D. Madge, L. Xing, T. Guo, A. Aspuru-Guzik, Nat. Biotechnol. 2019, 37, 1038–1040.
- Q. Gao, L. Yang, Y. Zhu, Curr. Comput.-Aided Drug Des. 2010, 6, 37–49.
- A. Yoshimori, E. Kawasaki, C. Kanai, T. Tasaka, Chem. Pharm. Bull. 2020, 68, 227–233.
- D. J. Weininger, J. Chem. Inf. Comput. Sci. 1988, 28, 31–36.
- A. P. Bento, A. Gaulton, A. Hersey, L. J. Bellis, J. Chambers, M. Davies, F. A. Krüger, Y. Light, L. Mak, S. McGlinchey, M. Nowotka, G. Papadatos, R. Santos, J. P. Overington, Nucleic Acids Res. 2014, 42, D1083–D1090.
- a) REINVENT: Molecular de novo design using recurrent neural networks and reinforcement learning https://github.com/marcusolivecrona/reinvent; b) M. Olivecrona, T. Blaschke, O. Engkvist, H. Chen, J. Cheminf. 2017, 9, 1.
- a) G. Wolber, T. Langer, J. Chem. Inf. Model. 2005, 45, 160–169; b) G. Wolber, A. A. Dornhofer, T. Langer, J. Comput.-Aided Mol. Des. 2007, 20, 773–788.
- J. Quan, T. Yahata, S. Adachi, K. Yoshihara, K. Tanaka, Int. J. Mol. Sci. 2011, 12, 971–982.
- S. Moll, A. Desmoulière, M. J. Moeller, J. Pache, L. Badi, F. Arcadu, H. Richter, A. Satz, S. Uhles, A. Cavalli, F. Drawnel, L. Scapozza, M. Prunotto, BBA Mol. Cell Res. 2019, 1866, 118474.
- S. Li, Z. Zhang, J. Xue, X. Guo, S. Liang, A. Liu, Med. Sci. Monit. 2015, 21, 2433–2438.
- P. Canning, L. Tan, K. Chu, S. W. Lee, N. S. Gray, A. N. Bullock, J. Mol. Biol. 2014, 426, 2457–2470.
- G. Poli, T. Seidel, T. Langer, Front. Chem. 2018, 6, 229.
- S. Kim, J. Chen, T. Cheng, A. Gindulyte, J. He, S. He, Q. Li, B. A. Shoemaker, P. A. Thiessen, B. Yu, L. Zaslavsky, J. Zhang, E. E. Bolton, Nucleic Acids Res. 2019, 47, D1102–D1109.
- G. R. Bickerton, G. V. Paolini, J. Besnard, S. Muresan, A. L. Hopkins, Nat. Chem. 2012, 4, 90–98.
- J. Wenzel, H. Matter, F. Schmidt, J. Chem. Inf. Model. 2019, 59, 1253– 1268.
- P. Ertl, A. Schuffenhauer, J. Cheminf. 2009, 1, 8.
相关资料
- LigandScout与REINVENT联用,实现深度增强学习从头设计TIE2选择性抑制剂
- 形状技术(OpenEye/ROCS)以及分子对接技术(OpenEye/FRED)与REINVET联用,实现增强学习从头设计
- 关于深度学习结构设计开源软件REINVENT
- 如果采样生成的结构打分分布不理想怎么办?
Yoshimori, A.; Kawasaki, E.; Kanai, C.; Tasaka, T. Strategies for Design of Molecular Structures with a Desired Pharmacophore Using Deep Reinforcement Learning. Chem. Pharm. Bull. 2020, 68 (3), 227–233. DOI:10.1248/cpb.c19-00625.
LigandScout是一款主打药效团的药物设计平台,支持基于结构与基于配体的药效团识别与虚拟筛选。如果您有蛋白结构、或复合物结构、或至少一个配体结构,理论上都可以生成药效团假设,然后与REINVENT联用进行从头药物分子设计。
Thomas, M.; Smith, R. T.; O’Boyle, N. M.; de Graaf, C.; Bender, A. Comparison of Structure- and Ligand-Based Scoring Functions for Deep Generative Models: A GPCR Case Study. J. Cheminform. 2021, 13 (1), 39. https://doi.org/10.1186/s13321-021-00516-0.
ROCS是基于形状的技术,理论上你只有一个已知的活性化合物就可以建立形状query进行增强学习训练。
而FRED分子对接技术则仅要求有一个蛋白,可以没有任何已知的活性化合物,就可以增强学习训练实现从头分子设计。
Ligandscout、ROCS与FRED等代表的药效团、形状与分子对接技术在增强学习中作为打分函数,而REINVENT则是深度增强学习从头设计的基础框架,由AZ开发、开源且免费。
Olivecrona, M.; Blaschke, T.; Engkvist, O.; Chen, H. Molecular De-Novo Design through Deep Reinforcement Learning. J. Cheminform. 2017, 9 (1), 48. https://doi.org/10.1186/s13321-017-0235-x.
Blaschke, T.; Arús-Pous, J.; Chen, H.; Margreitter, C.; Tyrchan, C.; Engkvist, O.; Papadopoulos, K.; Patronov, A. REINVENT 2.0: An AI Tool for De Novo Drug Design. J. Chem. Inf. Model. 2020, 60 (12), 5918–5922. https://doi.org/10.1021/acs.jcim.0c00915.
我个人的经验是:1)第2步用迁移学习生成模型代替用ChEMBL训练的先验网络;或者2)用集中库代替来自ChEMBL或ZINC的化学结构训练先验网络,此时先验网络自身就可以生成高质量的结构。
如何设计集中库,在博客里已经有非常多的算例了,比如:计算赋能工作流识别全新KRASG12C共价别构抑制剂。
我认为,集中库基本体现了药化专家对项目的理解,本身就是对药化专家的思维模拟。以集中库为训练集进行深度结构生成模型训练,可以进一步拓展、补充集中库,将传统集中库抽象为深度结构生成模型以实现对药化专家的模拟,并通过泛化能力实现对专家的拓展。最终,在结构采样步骤,深度生成模型被还原为集中库,并进一步用经典的虚拟筛选、各种过滤标准进行筛选实现最终的分子设计目的。
相关行业信息
- 日本英知德用Deep Quartet提供药物设计咨询服务
- 由Insilico Medicine发表的首篇采用深度学习结构生成设计化合物并被实验验证的文章
Deep Quartet是英知德的REINVENT+LigandScout的实现,技术流程如图6所示。更多Deep Quartet相关资料及其技术服务,请访问:Deep Quartet
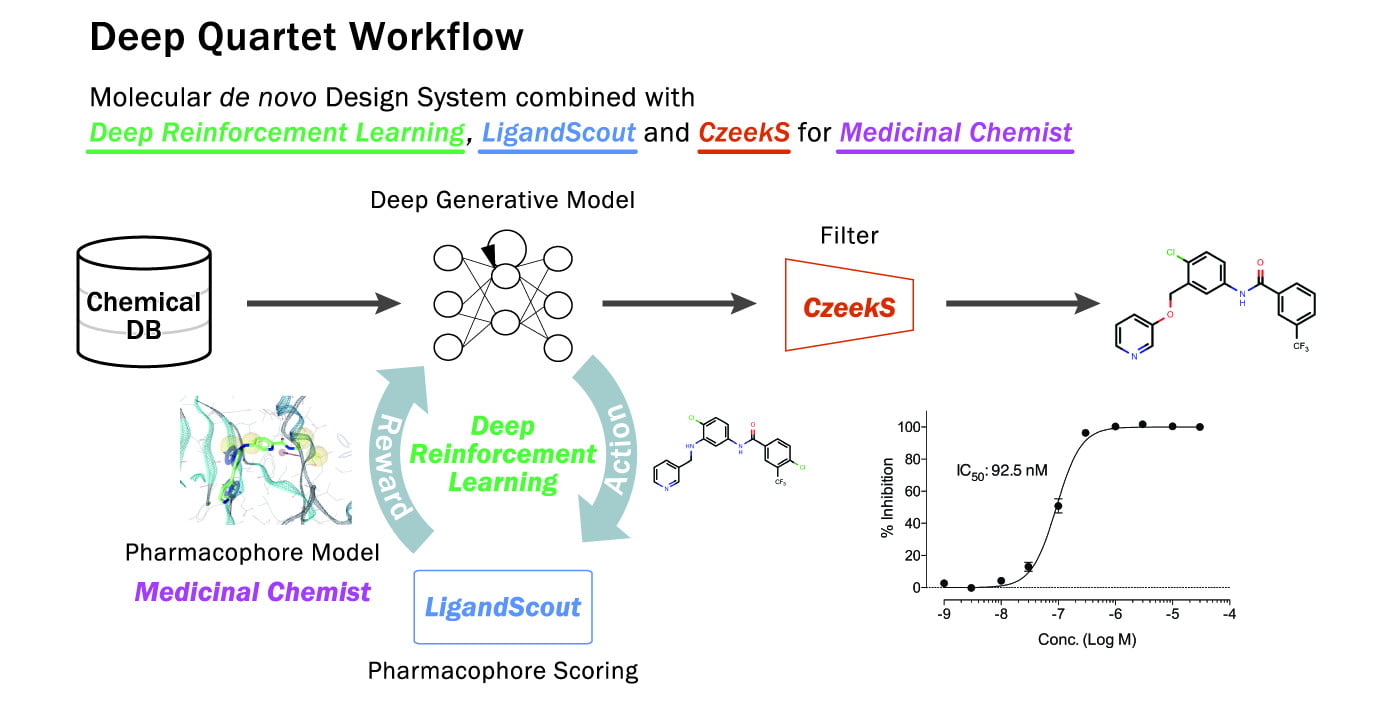
图6. Deep Quartet深度结构生成模型的技术实现
Insilico Medicine(英矽智能)是AI制药领域的领跑者,他们以DDR1激酶为模型体系,展示了深度增强学习在从头分子设计领域的应用潜力。
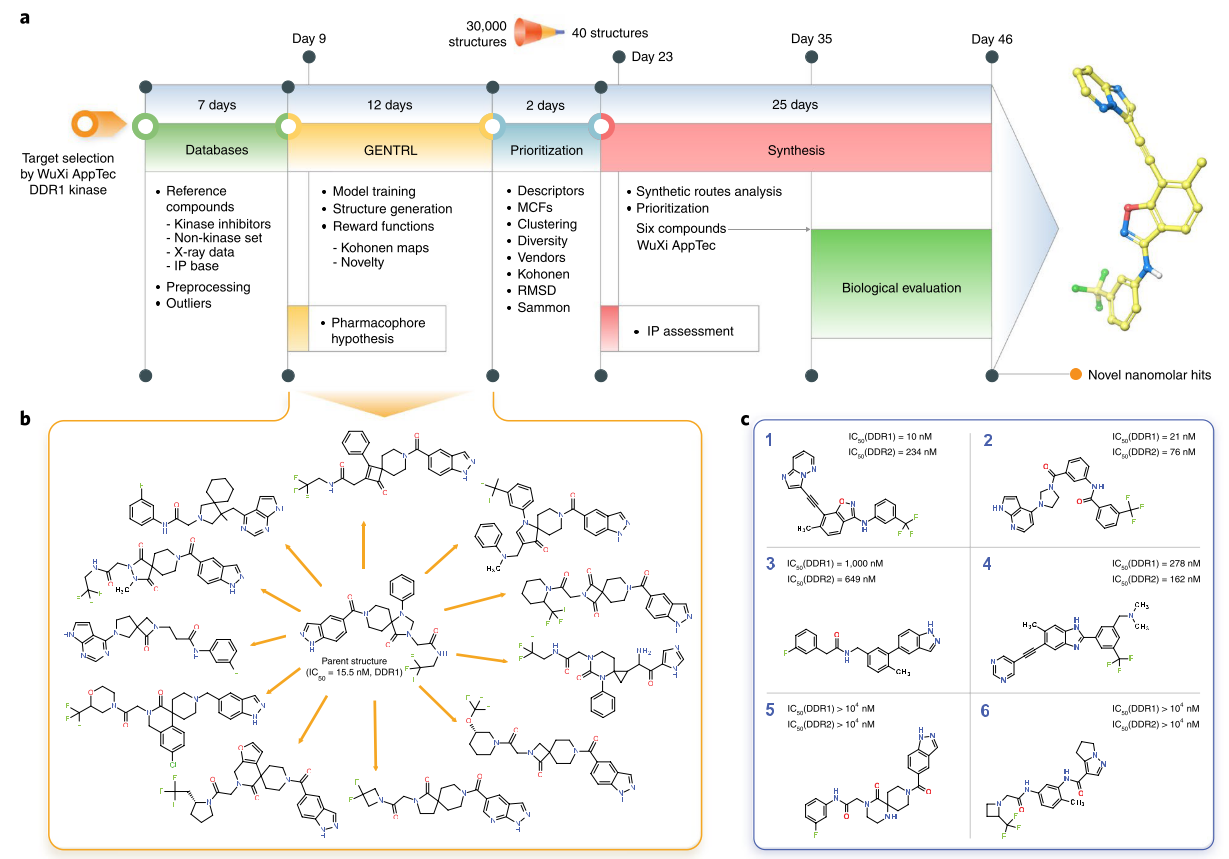
图7. GeNTRL模型的设计、工作流与nM苗头化合物。a,GeNTRL分子设计总的工作流与时间线. b, 已知的DDR1激酶抑制剂与生成的代表性结构;c, 生成的对人类DDR1激酶抑制活性最强的化合物
Zhavoronkov, A.; Ivanenkov, Y. A.; Aliper, A.; Veselov, M. S.; Aladinskiy, V. A.; Aladinskaya, A. V; Terentiev, V. A.; Polykovskiy, D. A.; Kuznetsov, M. D.; Asadulaev, A.; et al. Deep Learning Enables Rapid Identification of Potent DDR1 Kinase Inhibitors. Nat. Biotechnol. 2019, 37 (9), 1038–1040. https://doi.org/10.1038/s41587-019-0224-x.
获取一个月测试版license,亲自实践基于LigandScout与REINVENT深度生成模型
REINVENT为AZ发布的开源免费软件,想亲自实践基于LigandScout与REINVENT的深度生成模型,请联系我们获取1个月的LigandScout试用。
如果您需要帮助,我们可以提供有偿的深度生成系统部署与培训服务。可以帮您实现REINVENT与我们的LigandScout药效团技术、OpenEye分子对接软件OEDocking、形状技术ROCS、Cresset场点技术的Forge联合使用,进行基于药效团、分子对接、3D形状相似性、形状与场相似性的深度结构生成。


