
REINVENT与精确的结合自由能排序相结合的生成式主动学习
摘要:主动学习(Active learning,AL)是按顺序连续实验设计的一个具体实例,它使用机器学习来智能地选择下一个要评估的数据点或批次的分子结构。从这个意义上说,它可以紧密模拟实验室的“设计-制造-测试-分析”迭代循环(DMTA循环),以找到给定设计任务的最佳化合物。本文描述了一种结合生成式AI和基于物理的绝对结合自由能分子动力学模拟的AL协议,具体而言该协议使用REINVENT和ESMACS,用于发现两种不同靶蛋白3CLpro和TNKS2的新配体。作者在Exa级计算机Frontier上部署了该生成式主动学习(generative active learning,GAL)协议。结果表明,与基准相比,该协议可以找到打分更高的分子,在3CLpro案例里用ML模型代理对接打分,在TNKS2案例里用ML模型代理实验确定的结合亲和力。所发现的配体在化学上也是多样化的,并占据着不同于基准的化学空间。还通过改变每个GAL循环中进行自由能量评估的批量,以评估对GAL协议效率的影响,并推荐在不同场景下的最佳值。总的来说,该研究证明了基于物理的方法和人工智能方法相结合的主动学习的强大能力,这种结合可以在前所未有的规模下高效地采样化学空间,并直接适用于现代数据驱动的药物发现。

原文:Loeffler, H.H. et al. (2024) “Optimal Molecular Design: Generative Active Learning Combining REINVENT with Precise Binding Free Energy Ranking Simulations,” Journal of Chemical Theory and Computation, 20(18), pp. 8308–8328. Available at: https://doi.org/10.1021/acs.jctc.4c00576.
编译:肖高铿
1. 前言
为特定目的设计优化分子是化学的基础,也是发现新药和新材料的核心。实际上,这是一个迭代、缓慢且昂贵的过程1,2,通常通过DMTA(设计-制造-测试-分析)循环实现,在这个过程中,需要对新创造的化合物进行性质测试,并据此指导下一阶段的设计3,4。因此,人们自然地希望转向计算来以更快、更便宜地找到更好的化合物。
主动学习(Active learning,AL)是一种常用的机器学习(machine learning,ML)方法,它查询信息源,可能是人类专家(人机交互,human-in-the-loop)亦或是准确的计算预测器,以交互方式标记大量数据。AL算法试图找到一种最佳的方法来仅对大量未标记数据中的一个子集进行标记,从而加速学习过程。该子集是主动选择的,即智能地选择,以使得迭代次数最小化,并只将样本转发给专家,而算法预测这些样本将会增加知识或者不确定。构建预测模型的被动算法会随机选择子集,通常不是在迭代的方式下进行。因此,AL被归类为半监督式学习策略,因为专家只能明确地标记一小部分的数据集。将这些标签告知预测模型(代理,the surrogate),最终可以根据需要用于标记整个数据集。
人们已经注意到AL与贝叶斯优化(Bayesian optimization,BO5)的关系,特别是基于池的AL(pool-based AL6)。AL算法定义了一种高效的数据标记方法,并因此改善了预测模型;而BO则寻求找到未知函数(“黑盒”)的全局最优。在基于池的AL中,数据分布是未知的,因此只能查询离散子集的示例,而无法预先获取整个数据集。相比之下,在基于群体的AL(Population-based AL)中,数据分布已知,目标是找到分布的最佳密度7。本文将讨论一种旨在同时优化配体-蛋白质结合亲和力并保持高水平探索性的算法(即多样性以促进发现广泛的新分子)。
在分子设计中,AL通常利用大型数据库或供应商目录作为数据池8-10,此时,可以先验地知道数据集的大小。这些库的规模可以轻松达到数百万甚至数十亿个化合物11。在AL中,通常使用诸如对接或MD模拟之类计算成本高的方法来确定新的标签,例如结合自由能(也称为结合亲和力)或对接打分值作为亲和力的代理。用相对结合自由能(relative binding free energy,RBFE)方法(商业软件供应商经常将其称为“FEP”方法)高效优化结合亲和力的AL方法,最近才出现在文献中12-20,但它们也被用于优化RBFE协议本身17。将生成式AI与主动学习相结合的方法(GAL)最近才开始出现21,一个涉及肽类22的概念验证研究,一个是使用绝对结合自由能(ABFE,absolute binding free energy23)的应用性研究。后者专注于高保真替代建模,其中训练数据来自对接、BindingDB的实验结果24和一种双重解耦ABFE方法25。
AL依赖于oracle(注:指代人类专家或更昂贵的计算方法)通过直接观察获得的信息(而不是通过推断获得的信息)进行标记。创建代理模型(surrogate model)旨在重现oracle的预测,但其计算成本要低得多。这里使用的典型算法是在QSAR建模中使用经典ML方法,如随机森林(random forest)或状态向量机(state vector machine),但也可能是更复杂的人工神经网络(artificial neural networks)或高斯过程(Gaussian process),后者尤其经常与贝叶斯优化(Bayesian optimization)一起作为首选选择。然后可以使用代理模型来计算化合物库的一个更大的子集甚至整个库以生成标签,从而最终取代昂贵的oracle。然后用获取函数(acquisition function)——在BO中通常称为填充采样标准——以选择一个新的小子集用于用oracle评估。信息性、代表性以及多样性之前已被提议作为获取函数所需的条件26。代理模型(surrogate model)的质量和获取策略对于AL算法的设计至关重要,因为本研究的最终目标是找到优化分子的同时尽可能少地使用资源和/或时间。或者,最终的目标可能是构建最佳代理模型(surrogate model)以替代oracle来进行下一步的预测──例如,代理模型可以用作相关靶标的起始模型。
在本研究中,我们用生成模型代替了固定大小的库来实时创建分子——即从化学空间的分布描述中提取27,28。生成模型可以产生化学空间的一个巨大子集29,而供应商库自然仅限于包含在库中的分子,这些分子是受到其合成协议定义和限制的11。为此,我们使用REINVENT28的强化学习(reinforcement learning,RL)来生成受外部“信息”约束的最佳分子,这个外部信息即评估每个化合物适应性的打分函数。RL算法驱动具有通用化学知识的“先验(prior)”模型向着代表当前任务(目标)化学空间的专业模型发展。打分函数可以是一个几个打分组件的聚合函数(比如加权算术平均值或几何平均值)的。在此,代理模型是最关键的打分组件,因为它告知RL算法关于标签的信息,在本研究中,使用ESMACS方法(注:一种增强采样分子动力学与连续溶剂近似的MMPBSA)来计算靶标对化合物的结合自由能46,47。我们注意到计算社区通常将MMPBSA的结果称为“绝对结合自由能”,但它们的绝对值是非物理的,应该被视为打分值。还使用了其他次要的打分组件;这些将在方法部分进行说明。
我们的研究表明了,将强化学习与基于物理的方法相结合具有高效采样巨大化学空间的能力。该方法相对于以前类似方法的主要优势是:(a)用生成式AI(GenAI)生成高质量小分子,(b)在AL工作流中将GenAI与基于物理模型相结合,(c)在基于物理的oracle中使用了基于系综的模拟,这使得假阳性/阴性最少,(d)使用高达1000的批量(batch size)操作规模具有更好的综合性能,(e)在诸如如Frontier之类非常大的HPC平台上部署,和(f)短的挂钟时间。
2. 方法
本部分描述了RL与REINVENT的详细协议、ESMACS的模拟协议、GAL工作流程,以及在本研究中使用的蛋白质靶点的细节。
2.1 用REINVENT对化学空间采样
REINVENT的主要运行模式是强化学习(reinforcement learning,RL)28。通过这种方法,一个模型(代理)会迭代地偏向于靶标特征(target profile),该靶标特征描述了分子理想性质,通过打分组件的聚合函数来实现。在这里我们使用加权几何平均值将这些打分组件聚合为每个生成分子的一个总分。主打分组件是一个QSAR类似的响应模型(打分权重为0.6),它使用ChemProp (Version 1.5.2)30-33创建。ChemProp使用定向消息传递神经网络(D-MPNN)来产生性质预测模型。这个模型在这里作为一个替代模型(surrogate model),并且它的算法和超参数都是固定的。因此,我们假设该模型的预测能力将保持基本不变。在每次 GAL 迭代中,它会用一批经 ESMACS 模拟预测结合亲和力的新分子进行更新。3CLpro的初始模型基于~10,000个结构,它们通过一个替代对接模型找到34,35。这个ChemProp模型在经过5折、5系综(5个不同初始权重设置的模型)的交叉验证36后进行超参优化而创建的,其中最大迭代次数被设定为30。初始数据集按照0.8:0.1:0.1比例分割为训练、测试和验证集。在此步骤中以及所有的模型更新步骤中都使用了无特征缩放的RDKit 2D标准化操作32。在每个GAL步骤中,我们会根据第2.3节所述的方式更新模型。有关REINVENT协议的详细信息,请参阅参考文献28。
为了引导TNKS2模型,使用QSARtuna38处理了基准集37中的27个已知化合物和实验亲和力测量值,该方法允许从一系列经典机器学习算法中自动选择模型。最好的QSARtuna模型是一个随机森林模型,随后用于REVINVENT生成10,000个结构以进行ESMACS评估(在第2.2节中描述)。得到的结合自由能用于训练另一个ChemProp模型。初始模型是按照与3CLpro相同的创建方式创建,并根据2.3所述方式进行更新。
其他三个打分组件分别是QED39,立体化学,两者权重均为0.2,以及结构警报(structural alerts)作为过滤器起作用,这意味着与不希望的结构模式匹配的化合物将获得0或1的分值。QED为每个分子分配一个类药性打分。这是为了生成合理的类药分子并保持在ChemProp模型的应用域内,因为生成模型会非常迅速地优化以适应响应模型的弱点。例如,我们发现如果分子生成不受任何限制,REINVENT就会开始创建具有很长烷基链的分子,因为ChemProp模型对这些分子的打分很高。然而,这显然是不可取的。所有权重都在初步测试运行中手动调整,以平衡不合理化合物的产生和主要目标:生成具有更好结合自由能的分子。由于REINVENT先验(prior)不支持立体化学,因此带有立体中心的分子打分为0,以鼓励代理生成没有立体中心的化合物。结构警报是一组小的SMARTS模式,用于筛选和抑制不需要的子结构(官能团、环大小);有关详细信息,请参阅补充资料中的输入配置。
RL分为两个阶段运行28。第一阶段使用QED、立体中心和结构警报打分组件来创建一个合理的类药agent,以减少查询替代模型时对无用化合物的需求。这只需要在GAL协议的准备过程中进行一次操作。第二阶段则将这三个打分组件与ChemProp模型一起应用。只有在这个第二阶段中,在每个GAL步骤中才需要运行。对于分子生成,我们使用经典的Reinvent prior40,41,这是一种从头开始创建SMILES序列的新颖模型。每个GAL步骤中产生的新化合物的数量称为“批量(batch zise)”,其大小设置为100。作为学习策略,我们使用DAP42 ,其sigma值为128,学习率为0.0001。为了鼓励探索广泛独特的骨架范围,使用了多样性骨架过滤器。骨架是使用Bemis-Murcko算法43通过RDKit44实现生成的。多样性过滤器使用了一个包含10个骨架的memory size,并强制最小打分为0.7,这意味着具有特定骨架的分子在后续步骤中的打分均为零,前提是总分超过0.7。从RL运行的第二次出现开始,相同的SMILES被降级到零。起始记忆作为重播记忆45,其memory size为50,样本大小为10。对于3CLpro,我们将当前GAL迭代中打分最高的分子放入起始记忆;对于TNKS2,则将27个实验验证的结构放入起始记忆中。输入配置显示在支持信息中。
2.2 使用ESMACS进行结合亲和力预测
我们使用ESMACS协议46,47进行结合自由能计算。ESMACS基于MMPBSA48计算,但使用副本系综来获得具有稳健不确定性的可重复结合亲和力估计值。这是一种终点自由能计算法,在这种计算中,结合自由能是为结合状态(化合物-蛋白质复合物)和未结合状态(分离的蛋白质和化合物)之间的自由能变化。在这里,复合物结构、蛋白质和化合物的所有构象都从复合物的模拟中提取出来,这是终点自由能方法中常用的一种协议。这样的协议非常适合理性药物筛选项目,其中正确的结合亲和力排序比准确的结合亲和力计算更加重要。
2.2.1 模型的准备
从REINVENT生成的化合物首先使用FixpKa49(Version 2.1.3.0)进行处理,以在pH 7.4下获得正确的质子化状态。随后,使用OMEGA49,50(Version 4.1.2.0)为每个化合物最多产生200个构象。然后,用FRED49,51,52(Version 4.1.1.0)将准备好的化合物结构库对接到蛋白质上(PDB ID:3CLpro为6W63,TNKS2为4UI5)。具有最佳Chemgauss4对接打分的对接结合模式用于接下来的ESMACS模拟。
ESMACS模拟的准备和设置使用结合亲合力计算器BAC53进行。AmberTools23软件包54用于体系的设置以及结合自由能的计算。化合物力场参数使用GAFF2,蛋白质使用Amber ff14SB,水分子使用TIP3P模型,化合物的偏电荷用AM1-BCC方法产生的。蛋白残基的质子化状态是使用AmberTools54的“reduce”模块分配的。pdb文件中的所有水分子都被保留。所有的配体-蛋白质复合物被溶剂化在正交的水中盒子中,与蛋白质的距离最小为10 Å。添加了抗衡离子以平衡体系静电。
2.2.2 分子模拟
标准的ESMACS协议47每个化合物-蛋白质复合物使用25个副本进行精确预测。对于药物筛选项目,需要评估巨量的化合物。由于考虑到大量的配体、速度、节点小时数和低吞吐量而采用粗粒度协议55。在这里我们使用了一个由10个副本组成的系综,与标准ESMACS协议相比,盒子尺寸更小,并且缓冲距离为10 Å而不是14 Å。这种粗粒度协议不可避免地降低了预测精度,但非常适合高通量虚拟筛选55。NAMD356作为所有平衡和生产运行的MD引擎。对于每个单独的模拟,首先对蛋白重原子施加约束在初始位置上进行能量最小化。然后在50K下用Maxwell-Boltzmann分布独立生成初始速度,并将系统加热至并保持在300 K下长达60 ps。最后,每个副本进行了4 ns的生产模拟。在本研究中,我们分别针对3CLpro和TNKS2总共运行了约17,440次和约22,000次ESMACS计算,总模拟时间为约2毫秒。然而,我们要指出的是,所有的这些计算都是在世界上第一台Exascale机器Frontier上完成的,这使得我们可以同时运行这些计算以进行每次迭代。鉴于我们体系的MD模拟性能是每天150纳秒/单GPU(AMD Instinct MI250X),我们在每轮迭代中能够在50分钟的墙时间内获得整个化合物批次的结果。此外,我们注意到本研究构成了一个更大的工作流程的一部分,涉及多个额外步骤(将在其他地方发表)。
2.3 生成式主动学习
GAL循环的主要组成部分(见图1)包括ESMACS作为oracle,ChemProp作为代理模型,而REINVENT则实时生成分子并因此取代了更常见的固定大小的库12,14,57。在使用ESMACS评估当前批次后,代理模型将被更新。原则上,可以冻结ChemProp模型中选定的一层以避免“灾难性遗忘”(也称为灾难性干扰)58。然而,我们发现从每个GAL循环积累的分子集重新训练一个新的模型比选择性地冻结神经网络层产生了更好的结果。这当然需要更多的训练时间,但它也可能在一定程度上抵消协变量的变化。我们使用5折交叉验证和在每个fold中使用5个系综来创建代理模型。模型更新是从前一步的模型开始的,这样只需要不超过6个epoch(模型优化迭代次数)。但由于性能原因,我们只使用25个模型中的最佳模型,该模型由最小测试集RMSE确定,在所有构建的模型中基本上保持不变。然后用ChemProp模型作为自由能预测器与其它打分组件一起运行RL(如上所述),进行300-500次迭代。REINVENT形成了内部优化循环,其中RL用于分子优化,而外部循环是AL算法本身(见图1)。
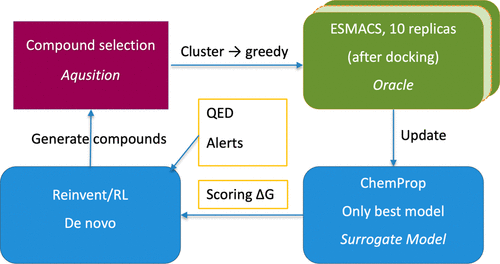
图1.整体工作流程的示意图。从右上角开始,ESMACS评估一组化合物的结合亲和力,然后将其与ΔGESMACS一起使用以更新预测ChemProp模型(右下角)。REINVENT使用经典的Reinvent prior招募这个模型以及其他打分组件(QED、Alerts、Stereo),生成数万个新的、潜在高分分子(左下角)。获取步骤(左上角)通过聚类选择生成化合物子集,并根据ΔGESMACS预测最高打分分子进行贪婪选择,将这些分子输入到下一个GAL迭代中。
GAL的主要参数是批量大小的选择和化合物进展的筛选标准。对于3CLpro,我们分别使用了250个和500个分子的批量大小。对于TNKS2,我们使用了五个批量大小:100、300、500、700和1000。从每个RL运行中获取化合物是在聚类后进行的,并且选择来自每个簇的最佳配体(聚类贪婪方法),正如Gusev等人15在2023年所做的那样。通过这种方式,我们促进了探索水平,这也得到了使用de novo REINVENT先验的支持。聚类是在将预测器空间减少到用UMAP59表示的二维之后生成的。我们采用了RDKit指纹作为描述符,并使用Tanimoto距离测量它们的相似性60。
我们在这里采用的聚类方法强调多样性,而对代表性的影响较小(通过选择每簇中最低的ΔG值),但它忽略了信息性26,即我们在GAL协议中不包括例如不确定性量化或信息增益。
2.4 测试靶标的描述
2.4.1 3CLpro
C30内肽酶,通常称为3CLpro或Mpro,是冠状病毒的主蛋白酶(main protease),在COVID-19 SARS-CoV-2生命周期中起着至关重要的作用。自2019年11月首次确诊病例以来,COVID-19于2020年1月30日至2023年5月5日由世界卫生组织宣布为全球性传染病15。从那时起,确认了数亿病例,并且数十万人直接死于该病毒61。 该病毒及其各种突变仍然是全球健康威胁。诸如瑞德西韦(Veklury)、奈玛特韦片/利托那韦片组合包装(Paxlovid)、巴克替尼(Olumiant)、托珠单抗(Actemra)和莫努匹韦(Lagevrio)等几种药物可用于治疗不同阶段和形式的新冠肺炎。然而,仍然需要找到新的活性化合物来应对不断变异的病毒和未来的相关疾病。第2.2节描述了用于模拟的3CLpro准备方法。
2.4.2 Tankyrase-2
从大型结合自由能计算基准数据集37中选择Tankyrase-2(基因TNKS2)作为又一个的测试算例。TNKS2不仅与致癌相关,并且调节各种细胞过程,如端粒维持、有丝分裂和葡萄糖代谢62。与3CLpro相比,选择它用于研究GAL在具有更封闭和受限结合口袋的靶标上的性能。它也是一个合适的测试算例,因为该系列同系物共有27个实验确认的配体可供使用。这些已知的配体用作种子结构来初始化GAL,以查看是否可以进一步提高结合亲和力以及生成的结构将如何与起始配体发生结构性差异。在第2.2节中描述了TNKS2的模拟准备。
3. 结果
在本节中,我们展示了GAL性能如何应用于两个不同的测试目标(3CLpro和TNKS2),从化学空间探索、生成化合物的结构多样性和化合物训练批量大小对获得结果的影响等方面进行了评估。
3.1 初步验证
为了检查ESMACS和对接的排序能力,我们对来自BindingDB数据库的1014个TNKS抑制剂进行了打分评估。请注意,ESMACS模拟是从对接结合模式开始设置并运行的。我们发现,Spearman’s rank相关系数ρ为0.33,Kendall’s counterpart相关系数τ为0.22;而对接的ρ=0.08,τ=0.05,这表明我们的对接协议不能为此体系的化合物排序。在本例中,ESMACS显示了相当适度的排序能力,但之前的研究已经证明ESMACS提供了准确的排名63-67。我们也指出,在一般情况下,不同来源(BindingDB包含多个出版物的数据自动聚合)的IC50不应合并进行相关性分析。然而,在这里,我们主要关注的是排序。实验和模拟中的误差仍然起着一定的作用。使用简单的蒙特卡罗方法,并假设ESMACS(10次重复)和实验IC50的标准误差呈正态分布,我们发现ρ=0.21,τ=0.14。pIC50的实验误差通常在0.1到0.3 log单位(0.14-0.41 kcal/mol)范围内,取决于实验方法、实验室等68。而在不同来源试验中,例如ChEMBL收集的数据集,标准偏差可能高达0.7 log单位69。在这里,我们假设了一个1.0 kcal/mol的误差以作演示目的。对于3CLpro的同等数据目前不可用,因为病毒,包括3CLpro的结合位点,正在不断突变,且数据集不一致。通过与每次AL步骤中随机选择化合物的方法比较,可以测试获取函数的有效性。图S14(3CLpro)和S15(TNKS2)展示了两种方法的自由能量分布。两个AL循环都从代理模型开始,使用最初的10,000个化合物(见第2.1节)。我们清楚地看到,随机获取比“智能”的聚类贪婪方法在寻找高分化合物方面效率低得多。
3.2 3 CLpro靶标
3.2.1 评估代理模型
用于预测结合亲和力的基于ChemProp的代理模型,初始训练使用了第一批10,000个结构,其结合自由能是通过ESMACS计算得出。这些初始结构根据它们从大型分子库中使用的代理对接模型得到的对接分数进行选择34,35。在后续迭代步骤中,那些从ESMACS计算中获得的化合物被添加到训练集中,并且在每个步骤中从头开始对代理模型进行训练。
在GAL迭代过程中,用于oracle评估的所有化合物的代理模型预测的结合自由能ΔGb值与ESMACS得出的值进行比较,并显示在图2、S2和S3中。结果分别显示了小(250个分子)和大(500个分子)训练批量的每个迭代步骤的结果。通常,代理模型的ΔGb值范围较窄是由于oracle获取过程所致,其中只有具有最低代理ΔGb值的化合物被发送到ESMACS oracle进行评估。
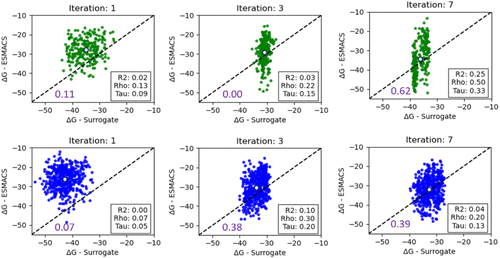
图2. 在选定的GAL迭代步骤中,针对3CLpro训练批量为n=250(绿色)和n=500(蓝色)时,代理模型预测ΔGb与ESMACS计算值之间的比较。每个图表的插图给出了R2系数以及Spearman和Kendall rank相关系数rho和tau。在一个迭代过程中所有代理模型预测和ESMACS计算的平均ΔGb显示为浅蓝色圆圈。所有的能量都以kcal/mol为单位给出。所有迭代步骤的结果见图S2和S3。代理模型的精度根据每个批量和迭代步骤给出,在这里将真阳性化合物定义为根据代理模型和ESMACS预测ΔGb \(<\) -35 kcal/mol的化合物。
根据结果,即使在初始训练后,代理模型开始具有合理的ΔGb化合物排序能力,但在第一次GAL迭代中发现其对新生成的化合物进行排序的能力相当有限。对于批量大小为250的情况,在GAL过程结束时恢复了大部分排名能力,而批量大小为500的情况下则恢复程度较低。
无论如何,在第一次迭代步骤中,代理模型已经识别出许多高分分子,根据代理分数,其中一些是真正的阳性。这些真正阳性的数量,即根据代理和oracle分数具有良好结合亲和力的化合物,从那时起稳步增加。真阳性与真、假阳性的和之比,即代理模型的精度,也稳步提高,如图2所示,并由ESMACS预测的ΔGb值逐渐接近最初由代理模型估算的过于乐观的ΔGb值所(浅蓝色圆圈)。对于较小和较大的训练批量的精度值分别从0.11和0.07开始,在第一个迭代后下降,最终分别上升到0.62和0.39,两个训练批量大小都有类似的趋势。鉴于固定模型容量和化学空间中的预期协变量偏移(参见下文的多样性分析)在主动学习中是一个主要问题,因此代理模型质量的初步恶化并不令人惊讶。随后恢复预测质量是由生成结构向AL过程结束时的结构收敛促进的。还值得注意的是,由于REINVENT prior不支持立体化学和电荷状态,所以代理模型建立在没有这些特征的化合物上。我们也没有包括一个描述蛋白质-配体相互作用复杂性的打分组件。在这里,REINVENT相对于ESMACS oracle操作的信息有限,必然会对oracle和代理(surrogate)预测之间的相关性产生影响。
在接下来的章节中,我们将看到即使替代模型对化合物进行排名的能力有限,但找到高分分子的精度足以产生具有越来越有利结合自由能的新化合物。
3.2.2 结合亲和力的分布
使用ESMACS估算的结合自由能ΔGESMACS分布如图3所示。此外,在图S5中,显示了在每个GAL循环后合并所有化合物打分最高的100分子的ΔGESMACS分布。
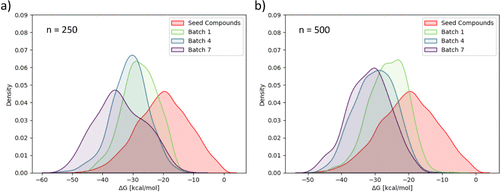
图3. 在3CLpro算例中,在选定数量的GAL迭代步骤后计算出的ΔGESMACS分布。(a)批量大小为250,(b)批量大小为500。用于训练初始替代模型的种子化合物的ΔGESMACS分布显示为红色。所有迭代步骤的ΔGESMACS分布如图S4所示。
总体而言,随着GAL的结束,能量分布向较低值稳定转移,并表现出收敛行为。与原始种子结构相比,低能尾部明显富集了更多高分分子。对于小训练批量,还可以看到低能尾部朝更低值延伸的现象。图S5中所有获得化合物的ΔGESMACS分布也显示,在整个GAL运行过程中,打分最高的100个分子的能量逐渐降低。
在图4a中,还显示了每个GAL周期后的ΔGESMACS分布。根据图3和4a,即使具有分布更相关的低能量尾部,但是观察到平均ΔGESMACS几乎线性的减少,在最后一个循环中也没有进一步向更低的ΔGESMACS移动。这意味着我们看到的是具有相似结构的局部ΔGESMACS最小值的富集,而不是找到结合亲合力更强的化合物。这种行为的一个可能解释是,GAL收敛到化学空间中的一个局部最小值,在那里结构变化无法使ΔGESMACS进一步降低。然而,我们也注意到预测的结合自由能低于-50 kcal/mol,这在我们的经验中是ESMACS自由能的下限。实现具有相对较低ΔGESMACS的结构多样性对于药物设计是非常有价值的,因为它增加了具有进一步优化可能性先导化合物的数量。
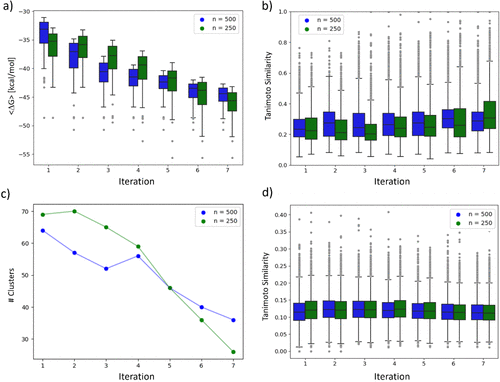
图4. 3CLpro学习批量大小为250和500的(a)ΔGESMACS分布、(b)Tanimoto相似性分布以及(c)每个GAL迭代中的结构簇数量分别以绿色和蓝色表示。(d)包含一个生成分子和用于初始替代模型训练的一个种子化合物的所有分子对的Tanimoto相似性分布。仅考虑了最低ΔGESMACS的100个化合物,并从每次迭代后的累积化合物池中提取这些化合物。
总的来说,结果清楚地表明,每个GAL循环产生的结构具有更强的预测结合亲和力,ΔGESMACS值相似或甚至比原始10,000个种子化合物中得分最高的分子低5千卡/摩尔。此外,在大约6-7个GAL循环后,化合物生成或多或少已经收敛了,即在每次循环之后只发现了少数几个相对较低ΔGESMACS的新化合物。
现在,人们可能会怀疑新生成的结构与原始种子化合物的最佳配体非常相似。因此,在下一节中分析了生成化合物的结构多样性。
3.2.3 结构多样性
图4b展示了每次迭代i后,从第0次到第i次迭代累积的化合物池中生成的100个最高分分子之间所有可能分子对的Tanimoto相似度分布情况15,这一分布直接与一组化合物的内部结构多样性相关70,60。两个化合物的Tanimoto相似度是基于它们的两个Morgan指纹71,72计算得出的。平均Tanimoto相似度在开始时非常低,大约为0.25,表明最初生成的结构具有高度多样性。随着GAL过程中后续循环化合物的加入,该值逐渐上升,但总体上保持在较低水平,普遍小于0.35。GAL过程中相似度的增加是化合物生成趋于收敛的一个标志,即一旦REINVENT学习到哪些类型的化合物属于高分分子,就会生成越来越相似的化合物。研究发现,无论GAL训练集规模大小,观察到的相似度分布都具有可比性。
为了更好地理解生成结构的多样性,使用累积的生成结构池对每个循环后的100个打分最高分的分子进行了聚类分析。采用Butina聚类算法73,并设定了0.5的Tanimoto相似度作为截断值。图4c展示了所发现簇总数的变化情况。观察到结构簇数量显著减少,这意味着随着每个GAL循环的进行,前100个分子可以被归入更少的结构簇中。这一发现与之前观察到的ΔGESMACS低能尾富集现象相一致,如图S5所示。
图S6展示了在不同数量的GAL循环后,所找到的每个聚类簇群体大小及其对应的平均ΔGb值,这同样是针对累积结构中打分最高的100个分子而言。在GAL过程中,正如之前所见,结构簇的数量减少,而化合物分散到各个簇中的范围收窄,最终只有少数几个簇最终具有明显的多的成员。每个簇可以被视为化学空间中的一个区域,在该区域内ΔGESMACS具有局部最小值。GAL过程在这个化学空间区域内用更多的结构逐渐填充这些区域,同时放弃ΔGESMACS更高的其他局部最小值。
为了更好地理解簇内和不同簇之间的结构相似性,图5展示了代表性化学结构。对于选定的簇,展示了具有最低ΔGESMACS的结构。每个簇的群体大小以括号形式给出。我们可以看到,Butina聚类生成的簇包含了具有不同骨架的结构,尽管各种结构模式频繁出现。总体而言,通过我们的GAL协议获得了一组具有不同骨架的高分多样化合物。
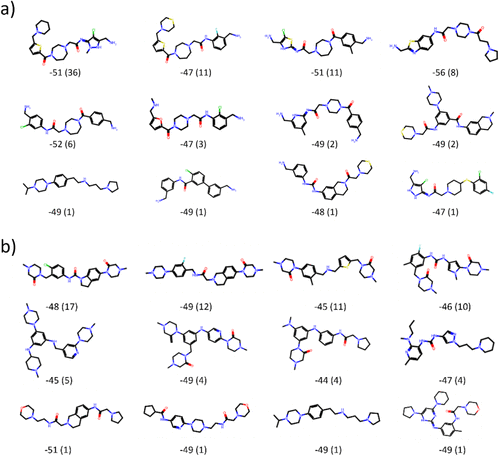
图5. 不同结构簇中具有最低ΔGESMAC的代表性化学结构。选择了八个最常见的簇以及另外四个具有最低ΔGESMAC值的簇。对GAL训练批量大小分别为(a) n = 250 和 (b) n = 500 的累积化合物池中的100个具有最低ΔGESMAC值的化合物进行了聚类分析,能量单位为kcal/mol,括号内给出了各簇的化合物数量。
接下来的问题是,找到的化合物与用于训练替代模型的原始种子化合物有多相似? 我们的GAL协议能够发现真正新颖的化合物还是仅仅发现了基于初始种子的变化?为了解答这个问题,评估了所有可能成对组合中包含100个生成的打分最高分子之一和一个原始种子化合物之间的Tanimoto相似性分布,并在图4d中显示了结果。
可以观察到,在GAL循环中生成的化合物与种子化合物之间的平均Tanimoto相似性低于0.13。最极端异常值在整个GAL过程中从约0.4降至0.35。这表明生成的打分最高分子确实是真正新颖的化合物,并且其结构与原始种子化合物有显著差异。此外,图4d中最大相似性的递减也表明,在GAL过程中,生成的化合物逐渐远离了由种子化合物占据的化学空间。为了进一步突出这一关键方面,我们在图6中通过降维可视化展示了化学空间的占据和漂移情况。
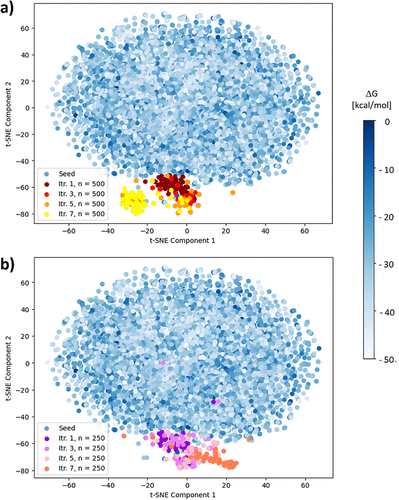
图6. 使用t-SNE将化合物的Morgan指纹投影到2D空间中,计算了所有种子化合物和来自大批量和小批量训练的所有生成化合物的3CLpro数据。用于初始替代模型训练的种子化合物根据计算出的ΔGESMAC值以不同的蓝色阴影显示(如右侧图例所示)。对于GAL训练批量大小分别为(a) n = 250 和 (b) n = 500 的精选迭代中,新生成分子与种子化合物的偏差用颜色编码表示(如图例中的插图所示)。这些分子是从每次迭代后累积化合物池中抽取出ΔGESMAC最低的100个化合物。
我们使用2D t-SNE74来可视化生成的打分最高的100个分子及其原始种子化合物的Morgan指纹在整个主动学习(AL)过程中的化学空间变化,如图6所示。蓝色数据点代表由原始种子化合物占据的化学空间区域。我们发现,在第一轮迭代后,新生成的化合物就已经显著偏离了种子化合物所在的化学空间区域。此外,我们也观察到高分分子的生成结构覆盖的化学空间比种子化合物的更小,这是因为大多数多样化的种子化合物也涵盖了广泛的结合亲和力范围。大约经过五轮学习循环之后,GAL(Generative Active Learning)训练批量较小和较大的结构移动到了相似但远离原始种子结构化学空间的区域,并且随后逐渐分裂成两个不同的化学空间区域。换句话说,在化学空间中,这两种GAL计算分别收敛到两个不同的局部ΔGESMACS极小值。这种现象并非完全由不同的批量引起,而是更多地反映了GAL协议非确定性的本质,该协议涉及使用各种随机种子和多概率采样的强化学习(RL)。最重要的是,这些结果清楚地表明高分分子的生成结构是真正的新化合物,而不仅仅是某些种子化合物的简单变体。
总体而言,我们证明了主动学习(AL)过程发现了真正新颖且多样的化合物,这些化合物具有不同的骨架结构和超出原始种子化合物中打分最高分子的结合亲和力。此外,在使用10,000个化合物进行替代模型初始训练后,对于大批量和小批量训练分别只需要7次迭代步骤(总共3500次和1750次oracle调用)就实现了这一目标。
3.2.4 配体结合模式的多样性
在图7中,我们研究了四个精选配体与3CLpro靶标蛋白的结合模式,以作为示例。这四个配体是在GAL运行过程中生成的,并且它们表现出很强的结合亲和力(−56 kcal/mol \(< ΔG_b < \)−49 kcal/mol),同时彼此之间具有较低的化学相似性。

图7. (a–d) 四个精选的具有高靶标结合亲和力预测值的配体与3CLpro结合口袋的结合模式。图(a,b)与图(c,d)中的分子分别取自训练批量为500和250的数据。各个化学结构及其结合自由能ΔGESMACS(单位:kcal/mol)在图上方给出。图中黄色高亮显示的配体结构是通过从10个ESMACS副本中抽取4 ns分子动力学(MD)模拟快照后叠加得到的。蛋白质表面根据表层原子类型进行着色展示。每个配体及其附近蛋白质在图右侧以更详细的视角呈现。
我们发现这四个配体有不同的结合模式,这一点通过配体与蛋白质之间接触面积的不同以及由此产生的配体-蛋白质相互作用的显著差异得到了证明。对这些复合物进行更详细的检查表明,配体胺基和蛋白质之间氢键相互作用的重要性,在生成的配体中许多强结合亲和力的分子都含有此类氢键,而配体羰基似乎较少参与氢键形成。在比较不同配体时,配体胺基与不同的蛋白残基形成了不一致的氢键,仅与阴离子Glu166和Thr25之间形成了一致的氢键。此外,我们发现配体与活性位点的不同部分发生相互作用,在图7c中所示的唯一非线型化合物的额外环状基团与活性位点特定区域发生专一结合,而这一区域是线型化合物所没有的。
更全面地分析结合模式超出了当前的研究范围。然而,总体而言,观察到的结合模式表现出额外的多样性,这在后续研究中可能通过组合与结合口袋不同部分相互作用的片段来加以利用。
3.2.5 计算效率
比较不同训练批量生成的GAL化合物的质量,如本研究中讨论的那样,一个问题是哪个批量在计算上最有效。在GAL工作流程中,最具计算挑战性的步骤是为每个oracle调用ESMACS计算 ΔGb值。虽然批量(batch size)越大需要的计算资源越多,但正如之前所述,找到高分化合物所需的迭代次数越少。这种行为可能有利于使用大量计算节点并行运行的大批量的计算;然而,在没有足够计算资源的情况下,较小的批量可能效率更高。
为进一步研究这个问题,我们将计算效率η定义为每进行一次oracle调用所找到的合适结构的数量:
$$
𝜂={𝑁_{CG,Δ𝐺_{max}} \over n_{oracle}}
$$
oracle调用的次数(noracle)简单地等于训练批量(batch size)与迭代次数的乘积。定义生成的化合物何时被认为是合适的则较为复杂。一般来说,一个好的化合物应具有较大的结合亲和力,并且其结构与其他已生成的化合物在结构上应有足够差异。我们定义当化合物的结合自由能\(ΔG_b < ΔG_{max}\)时为合适化合物,其中考虑了不同的阈值ΔGmax。此外,还对所有\(ΔG_b < ΔG_{max}\)的结构使用Butina算法进行了聚类分析,并计算出簇数量作为找到的不同化合物组的数量NCG,组中的每个化合物与其他组相比在结构上足够不同。聚类算法的相似度截断值scutoff是两个结构属于同一簇所需的最小相似度。较大的scutoff值意味着只有非常相似的化合物才会被分到同一个簇中,并且生成的簇数量较少而规模较大,每个簇在化学空间中的覆盖范围更大。
在使用较小的scutoff值的情况下,化合物被分组到更多的簇中,并且每个簇中的成员数量较少。同时,要求更严格的ΔGmax为−40 kcal/mol,这意味着我们正在寻找具有高结合亲和力的化合物,但结构多样性不那么重要,因为所有新生成的化合物都计算在η中,无论它们与已生成的结构有多相似。这种情况类似于药物发现中的先导化合物优化。相比之下,在更宽松的ΔGmax = −35 kcal/mol和较大的scutoff情况下,强调的是结构多样性和化学空间的覆盖范围,而不是结合亲和力,这通常适用于更具探索性的苗头化合物发现场景。因此,通过选择ΔGmax和scutoff,我们可以评估GAL在这两种不同情况下的有效性。
对于两个不同的训练批量(batch size)、两个不同ΔGmax值以及不同的scutoff,整个3CLpro GAL中的η结果如图8所示。ΔGmax = -35 kcal/mol大致对应于最后一次GAL迭代后获得的ΔG分布的最大值(参见图3),而ΔGmax = -40 kcal/mol代表了一个更严格的标准来定义合适的结构。在所有情况下,较小的训练批量(batch size)n=250计算更加高效。对于scutoff = 0.3,在增加迭代步骤时观察到效率的改善,而对于scutoff = 0.7,在整个GAL运行过程中效率保持相对稳定。
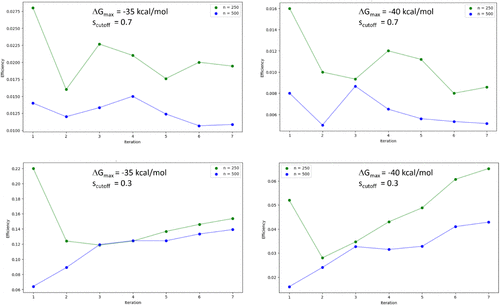
图8. 在3CLpro靶点算例中使用两种不同的训练批量(batch size)在每次迭代步骤之后的GAL效率。效率定义为在给定迭代步骤中所有累积生成结构的所有oracle调用中发现的结构簇的数量。仅考虑\(ΔG_b < ΔG_{max}\)的配体。聚类采用Butina算法进行,并根据各自图表给出相似度阈值。
3.3 Tankyrase-2靶标
3.3.1 生成第一个批次的化合物
tankyrase-2(TNKS2)靶点的GAL起点是一系列实验确认的结合自由能已知的27个配体,范围从−35到−26 kcal/mol。使用ESMACS计算结合亲和力与实验测量的亲和力进行比较,如图S7所示,在该图中通过模拟获得了合理的化合物排序。然后我们按照第2.1节所述的方法生成了第一个训练批次的化合物。接下来,我们将使用最初生成的化合物进行的GAL步骤称为迭代0。进行了五次GAL步骤,仅在每次迭代时改变训练批量(batch size),分别为n=100、300、500、700和1000个化合物。总的来说,我们发现只需四次迭代就足以使结果收敛,如下所示。
3.3.2 代理模型的质量
在图9中,针对一系列不同批量(batch size)和迭代步骤(图S8包含整个数据集的数据),对ChemProp替代模型(surrogate model)的结合亲和力预测值与ESMACS计算的ΔG值进行了比较。首先,我们获得了比3CLpro算例更好的替代模型预测质量。获得较大的Spearman ranking和R2系数,分别大于0.7和0.6。对不同的批量进行比较可以发现,可靠的替代模型都是在第3轮迭代后被发现,除了最小训练批量n=100的情况除外。我们也观察到对于n=500及以上的情况,良好的替代模型已经在第一轮迭代步就被发现了。对于GAL来说这意味着根据ESMACS找到了更多的真阳性,即具有有利结合亲和力的化合物,并且精度更高。接下来描述这将如何影响GAL的整体情况。关于相比3CLpro而言,在替代模型质量更好的背后可能的原因将在第3.3.5节讨论。

图9. 在TNKS2算例中,当选定GAL迭代步骤、训练批量为100、500和1000分子时,代理模型预测的ΔGb与计算的ESMACS值之间的差异。每个图表的插图给出了R2系数以及Spearman与Kendall等级相关系数rho和tau。所有代理模型预测值和ESMACS计算值在一次迭代中的平均ΔGb显示为红色圆圈。所有的能量都以kcal/mol为单位给出。所有训练的批量和迭代步骤的结果如图S8所示。
3.3.3 ΔGb值的分布
图10显示了选定迭代步骤获得的ΔGb值分布(完整数据集包含在图S9中)。出于比较的目的,还展示了原始27个配体同系物系列的实验值以及从第零次迭代开始化合物的ΔGb分布。我们发现,在第一次迭代后,与初始第零次迭代化合物样本相比,已经取得了显著改善,并且分布的高亲和力尾部甚至延伸到实验确认配体的结合能之外,这些配体本身就已经具有很强的结合亲合力。此外,我们发现对于n=500及以上的情况,第一步之后的迭代几乎不再进一步改进能量分布,而对于较小的批量n=300,通过额外的迭代仍然可以实现改进。对于n=100的情况,更多的迭代并没有带来太多的改进,而高亲和力尾部向更低的能量方向扩展的程度也比其他批量要小。

图10. 在TNKS2算例中使用不同批量的GAL迭代的ΔGESMACS分布。用于训练初始替代模型的10k种子化合物的ΔGESMACS分布显示为绿色,作为批号0。出于比较目的,27个化合物的实验ΔGb分布用红色显示。全部批量和迭代步骤的结果见图S9。
总的来说,ΔGb分布的收敛在单次迭代中基本快速实现(n=300除外),而在3CLpro算例中则需要五次或更多次迭代。图11a显示了GAL运行过程中累积生成化合物的打分靠前的100个分子的ΔGb分布变化:随着批量的增加,\(<ΔG_b>\)值趋于降低,并且随着迭代步骤数量的增加而稳定下降。在这里,较大的批量能够开发出更精确的代理模型,如前所述,这有助于\(<ΔG_b>\)的适度改善,但这也伴随着更多的oracle调用的计算成本。GAL的计算效率将在第3.3.6节讨论。
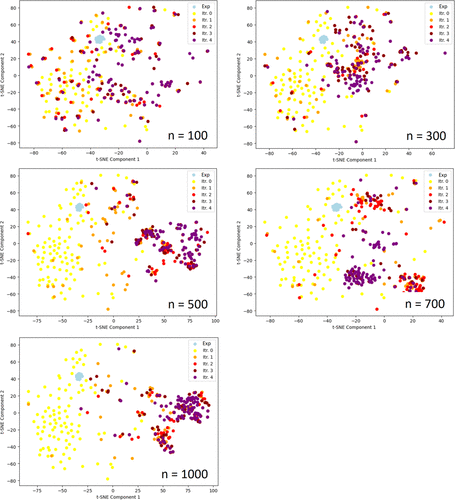
图11. TNKS2算例的(a)ΔGESMACS分布,(b)Tanimoto相似度分布,以及(c)每个GAL迭代和不同学习批量的结构簇数量。(d)显示了一个生成分子和一个用于生成第一个训练批次化合物的27个同系物系之一的所有分子对的Tanimoto相似性分布,其中生成的分子从每次迭代后累积的化合物池中取出,仅考虑ΔGESMACS最低的100个化合物。
我们还探索了通过在替代模型中增加来自BindingDB24的已知TNKS2 IC50值,是否可以进一步提高结构多样性。将与27个基准化合物具有相同的母核且\(IC_{50}≥1μm\)的结构去除,留下484个配体。从这些配体中随机选择20个,并将其添加到迭代3中的80个化合物以继续GAL,训练批量100。结果ΔGb分布如图S11所示。我们发现,与没有融合BindingDB的GAL相比,在经过第2次迭代后,ΔGb分布有轻微改善,但在5次迭代后没有获得任何改进。
融合了BindingDB化合物样本训练的替代模型的质量如图S12所示。结果与之前在图9中显示的训练替代模型相当。平均Tanimoto相似度为0.16和0.15之间,这基本上与以前观察到的没有融合BindingDB化合物的GAL相同,见图11b。总的来说,似乎额外融合BindingDB结构并没有提高GAL的结果质量。
3.3.4 结构多样性
生成结构的内部结构多样性如图11b所示。一般来说,结构的多样性往往比3CLpro算例中的更大。平均Tanimoto相似度随着迭代的增加而增加,除了训练批量n=100外,这表明在化学空间中达到收敛。较大的训练批量获得较小的多样性,在这种情况下可以假设在ΔGb最小值附近的化学空间更全面地采样导致了更多相似的结构。
图11c显示了,使用Tanimoto相似度截止值为0.5时的Butina算法,从累计生成的结构中选出的打分最高的100个分子结构的簇数量。此外,图S10显示了每个结构簇的平均ΔG及其群体大小。对于3CLpro算例,我们看到,在GAL过程中,随着与具有低ΔGb能量最小值相关结构的簇逐渐增多,而与较高ΔGb最小值相关的簇减少,群体数量显著下降。另外,使用较大批量进行的GAL导致簇数量较少,这再次与化学空间中的更明显的收敛有关。
图12显示了一些生成的化合物结构,这表明生成的具有良好结合亲和力的化合物涉及多种不同的骨架。考虑到生成化合物的初始样本仅基于共享相同喹唑啉酮骨架的小同系物,这一点尤其值得注意。然而,我们还发现预测的配体中经常出现喹唑啉酮及其类似骨架(见图12)。当直接与同系物进行比较时,GAL生成的结构与那27个已知配体相比确实表现出较低的Tanimoto相似性,大多数值低于0.3,如图11d所示。我们还观察到,在较大的训练批量下相似度分布向较大值伸展较少,即,使用的GAL训练批量越大,与原始分子的差异也越大。
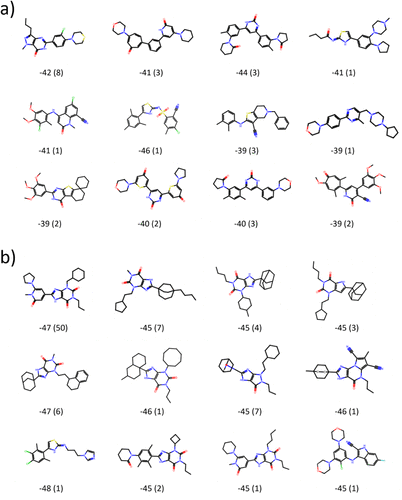
图12. 在TNKS2算例中从不同结构簇中精选的具有最低ΔGESMAC的代表性化学结构。选择了8个成员数最大的簇以及4个具有最低ΔGESMAC的簇。聚类分析是对在每次迭代后的化合物累积池中打分最靠前的100个化合物进行,用于GAL训练批量(a)n=100和(b)n=100。括号中数值是簇的成员数量。能量单位为kcal/mol。
这一趋势符合训练批量增加而平均ΔGb值下降,这对应于化学空间进一步收敛,从而导致在批量n=1000的训练中化学空间包括如腈和桥环等官能团。
在第3.3.5节中我们将展示这些基团如何对ΔGb最小化做出贡献。除了改善ΔGb之外,从如图S14中我们还观察到批量越大则QED打分越高,即更类药性越好,尽管我们在图12b中也看到了一些不太常见的化合物片段,但它们通常不会出现在典型药物中。
此时,重要的是要注意到许多生成的化合物虽然具有有利的ESMACS结合亲和力ΔGb和QED打分,然而在药物发现项目中一些化合物不太可能被选择用于进一步开发,因为它们可能太难合成或缺乏其他特性。这些问题的改进不在本研究的范围之内。
图13用2D t-SNE可视化了在GAL过程中不同训练批量所穿越的化学空间。我们观察到原始的27个配体(显示为浅蓝色数据点)集中在非常小的区域内,而生成的10k结构(显示为黄色)则覆盖了一个更大的化学空间。在整个GAL过程中,可以观察到向与迭代0中的结构所覆盖的空间不同的化学空间收敛的趋势,这种趋势对于具有较大化合物训练批量的GAL更为明显。对于n=100和300的情况,似乎尚未发生收敛。此外,我们还发现当n=500和n=1000时,收敛到了相似的空间区域;而对于n=700,则GAL收敛到了一个稍微不同的区域。
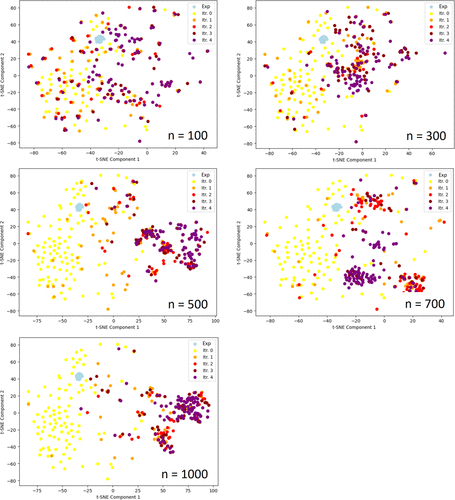
图13. 在TNKS2算例中合并所有批次化合物,然后使用t-SNE将Morgan摩根指纹投影到二维空间以进行化学空间的比较。第0轮迭代的化合物用黄色表示,指的是从最初的10,000个化合物中提取的分子。实验测得结合自由能的27个配体用浅蓝色显示。生成的分子按图例嵌入的颜色编码。所示分子是从每次迭代后积累的化合物池中选出的ΔGESMACS最低的100个化合物。
总的来说,本小节中报告的发现表明GAL可以生成具有高内部多样性的结构,与起始的27个配体有明显的区别。结果还表明,在训练批量\(n≥500\)的情况下,根据图13和S10所示的结果,最后一轮GAL迭代后产生的前100个结合亲和力最佳化合物可被分为较少的结构簇,并且ΔGb较低,即所产生的打分最高的分子占据化学空间中的更窄区域。这也说明了批量越小的GAL可能需要月越多的迭代步骤以便从结构收敛和ΔGb优化获益。
3.3.5 配体结合模式的多样性
图14显示了四个代表性的、与TNKS2结合的低ΔGb结构。与3CLpro算例相比,一个明显的定性差异是:在3CLpro算例中,由于靶标的开放结合口袋较大,发现了多种不同的结合模式;而在TNKS2算例中,结合口袋狭窄且封闭,这导致了存在一个明确的结合模式,并且尽管配体之间存在较大的结构差异,在结合口袋中的配体构象变化要少得多。对配体-蛋白质界面进行更仔细的研究表明,一些改善ΔGb的基团经常出现在生成的结构中。例如,通过三键将腈基引入到配体中,以优化配体和蛋白质表面之间的距离。桥接双环基团被证明可以作为定制的“塞子”,从一侧关闭活性位点的开口,从而使得与蛋白质范德华相互作用最大化,否则很难用平面环或其他基团实现。这些例子说明了REINVENT如何优化GAL中化合物的设计,使其适合蛋白靶标的活性位点。
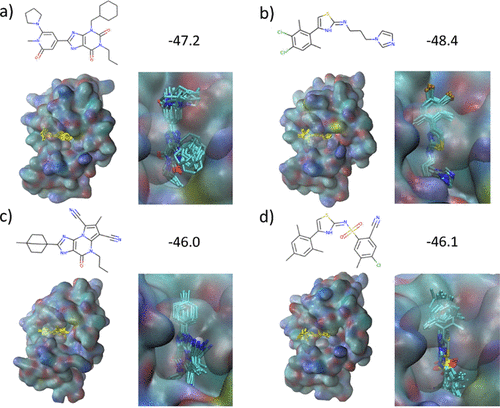
图14.(a-d)在TNKS2结合口袋中预测具有高结合亲和力的四个精选配体及其结合模式,分别来自最大和最小训练批量,分别为1000个(a、b)和100个(c、d)。化学结构及其结合自由能ΔGESMACS(单位为kcal/mol)显示在图像上方。每个图像左侧突出显示的黄色配体结构是每组十个ESMACS副本在4ns MD模拟后快照的叠加。蛋白质表面根据表面原子的类型进行着色显示。右侧更详细地展示了每个配体与其蛋白附近的结构。
可以合理地假设,在TNKS2活性位点中的受限空间和相应定义良好的结合口袋结构,使得在REINVENT中表示为1D SMILES代码的配体结构与该活性位点内配体的三维对接结合模式以及由此产生的结合自由能之间存在比较清晰的关系。这可能有助于开发比3CLpro算例更具预测性的GAL替代模型(因为我们的替代模型只考虑配体结构而不考虑蛋白质结构,因此没有明确捕捉蛋白-配体相互作用),从而能够在较少的GAL步骤内发现具有良好结合亲和力的新结构。这表明尽管对于所选测试靶标都取得了成功,但GAL过程的表现仍取决于靶标的活性位点对配体的限制程度。
3.3.6 计算效率
图15显示了不同训练批量、两个ΔGmax值以及不同scutoff时,TNKS2算例的计算效率η的结果。ΔGmax = -35 kcal/mol描述了大约是经过GAL后获得的ΔGb分布的最大值,而ΔGmax = -40 kcal/mol是更严格的选择合适化合物的标准。
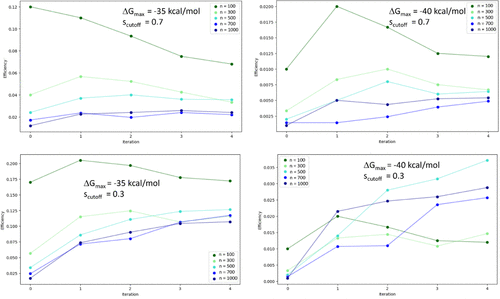
图15. 在TNKS2算例中,使用不同训练批量,在每个迭代步骤后的GAL效率,颜色编码如图例所示。效率定义为在给定迭代步骤中找到的所有累积生成结构的oracle调用次数。仅考虑\(ΔG_b < ΔG_{max}\)的配体。聚类使用Butina算法进行,Tanimoto相似度阈值见相应图表。
根据这些结果,我们观察到在迭代步骤增加的情况下,批量越大(n=500或更大)效率越高;而对于n=100的情况,效率保持不变或下降。这一发现与图9所示的代理模型质量一致。对于\(n≥500\)而言,更精确的代理模型能够更好地识别合适的新型化合物,从而提高了GAL过程的效率,但代价是需要更多的oracle调用次数。事实证明,在大多数情况下,批量越小效率越高,尤其是最小的批量n=100。只有在ΔGmax=-40 kcal/mol和scutoff=0.3的情况下,才发现了n=500的批量是最高效的,其次是n=1000和n=700的批量。如第3.2.5节所述,这些参数定义了η,它更适合于优先级排序以寻找结合亲和力大的化合物而不是化学空间探索的情况。
这些发现表明,探索性的GAL受益于较小的训练批量,而开发性则会以较大的批量而更高效地运行。这一发现与在3CLpro算例的情况有所不同,在3CLpro算例中,较小的训练批量总是导致更高效的GAL。这种差异的原因可能是,比之TNKS2算例,在3CLpro算例中代理模型的精度在所有情况下都要差。似乎想要提高GAL效率,需要一个足够精确的代理模型。总的来说,我们的研究结果表明,在大多数情况下,当代理模型的质量事先未知时,小批量是更高效和更安全的选择。
4. 讨论
在本文中,我们证明了主动学习(Active learning,AL)可以是一种高效的方法来改善特定靶标化合物的优化。重要的是,我们指出了如何遵循行业传统来使用“主动学习”12-16,18-20。然而,在引言中我们还指出了,我们的重点不是对创建一个代理模型以最优地找到新的标签即结合亲和力而感兴趣。事实上,在我们的AL应用中,这些模型(代理模型和RL代理,后者实际上是次要的代理模型)仅仅是人为创造的,而重点是产生足够数量ESMACS计算高分的分子。我们并不是真正试图在严格意义上的“优化”中寻找函数的最大值。图S1显示了在AL运行过程中发现高分分子的情况,并且我们可以看到,在任何RL循环以及任何AL步骤中都可以非常早地发现高分分子。这对于预算有限的情况下非常重要,因为项目只有有限的时间或资金可用。正如我们所展示的那样,生成式主动学习(GAL)完全有能力在这样的范围内找到高质量的化合物。我们注意到RL本质上具有高度随机性,不同的超参可能会影响化学空间探索。但是,这意味着重复运行可能会导致生成分子的更具多样性。由于计算成本和预算限制,多次重复运行可能是不可行的。今天的药物发现仍然很大程度上是一个“数字游戏”,意味着需要生成足够的建议(创意),然后由药物化学家分别评估。我们也强调,我们在这里只使用了数量非常有限的打分组件,远远不足以评估化合物作为可行候选药物的质量。当前的设置忽略了分子的开发潜力,例如合成可行性、PK/PD、ADMET、IP、制剂、理化性质等等。从这个意义上说,目前的工作只是一个概念验证,为了证明GAL可作为一种高效的资源节约型工具用于分子优化。因此,在未来有必要努力实现更现实的靶标概述(target profile)。本研究使用的打分组件包括结合自由能预测(主要的先验知识)和两个打分组件(QED,chemical alerts),这是非常简化的模型,用来约束产生的化合物。在本研究中,我们完全没有考虑合成可行性,虽然在实际分子设计中,显而易见的,这是一个主要的设计目标。我们还补充说,一些生成的结构,如图5所示,可能不是化学稳定的,因此需要仔细的后处理或者需要如这里讨论的进行更加复杂的合成可行性评估。因此,不出所料,用GAL探索的化学空间原则上不能期望与实际上合成并经实验确认的配体相似。出于同样的原因,生成的高分分子可能不会表现出与之前报道的配体太多的相似性,但这对基于新发现的化学空间的新分子设计是有益的。在未来,我们计划使用诸如AiZynthFinder75之类地内部解决方案以及其他方法来解决合成能力和生物学问题。
我们还证明了将基于配体的生成模型和基于物理的方法相结合的必要性。人们已经证明,基于结构的方法显著提高了生成模型的效果76-78,并且证明了在低数据量条件下基于物理的MD描述符为何是必要的79。对接本身通常与实验或更准确的基于MD的方法相关性较差,并且我们在本研究中也观察到了这一点(见图S13)。因为对接作为打分函数往往不够精确80,81,所以对接往往用于富集和预筛选。这本文中,我们使用基于物理的方法ESMACS通过代理模型来告知RL代理(RL agent),并展示了这是如何驱动我们的GAL工作流以便高效地产生更好的结合分子。我们的研究还表明了GAL工作流还有许多地方有待改进与完善。我们通过具有固定超参数和几何形状的深度学习模型构建了一个固定的静态代理模型。原则上,我们必须搜索整个模型空间才能找到与预测结合自由能相容的模型,其与基于物理的结合亲合力预测相一致。我们注意到这个模型完全是基于配体的,没有考虑蛋白结构或配体-蛋白相互作用。早期将一些基于结构的信息纳入主动学习的努力只显示出有限的效用13。先前的RBFE研究利用了自动QSAR建模15,17,这一点我们可以利用自己的QSARtuna软件38来实现。然而,用ChemProp建立一个模型是相当耗时的任务,特别是在使用多折交叉验证和构象系综的情况下。根据数据和数据大小的不同,在主动学习过程中可能需要改变模型构建算法以找到最佳模型,例如,在TNKS2算例中,对于27个配体这样相对较小的数据集,我们使用了随机树模型;而在实际的主动学习计算中,我们选择相当大数量的化合物作为种子,这时侯使用神经网络模型就更为适合32。我们也在每个主动学习步骤中从“零开始”创建代理模型,即从累积的化合物数据集中重新开始,而权重和偏差是从上一步的AL模型中获得的。但是,我们也可以使用无层冻结技术(layer-freezing techniques)32来加速代理模型的训练。同样,RL代理(RL agent)也在每个主动学习步骤中被重新培训;我们可以检查前一个主动学习步骤中的最终或后期阶段的代理是否可以用作下一个GAL周期的合适起点。这需要权衡对模型可塑性82-84的担忧,其中协变量的变化,在这里表现为化学多样性的变化,是一个值得关注的问题。虽然主动学习不是连续的学习,但即使是在时间上的有限数量的代理进展步骤中,这也可能会成为一个问题。
在本研究中,我们进行了虚拟筛选或虚拟苗头化合物发现练习。经典的REINVENT prior方法允许从头设计,这意味着分子生成不受结构限制,但仅受打分组件的约束。在这个意义上,我们在进行逆向设计,即通过描述所需的性质空间来创建新分子。对于实际先导化合物发现,特别是与AL和自由能模拟相关,通常是在一个公共母核或有限数量母核周围开发新的化合物,这些母核可以通过RBFE映射网络连接起来。为了促进这一点,我们可以使用Mol2Mol模型85,该模型基于分子对相似性的关系限制分子生成,例如使用Tanimoto相似性或匹配的分子对。这将使我们能够根据相似性标准的严格程度来对给定的骨架本身进行修饰。如果需要骨架约束,则可以使用我们的Libinvent模型86,因此可以在固定、预设的母核周围产生新的分子。目前,REINVENT并不直接支持基于片段的方法,也就是说,我们的Linkinvent模型87只支持两个“弹头”(片段),然后可以用单个片段将它们链接起来。这可以扩展到允许对多个片段连接。例如,3CLpro靶点有一个相当大且分支状的结合位点(见图7),所以可以在不同的结合位点位置上连接多个已知片段,如本研究所发现的那样。
ESMACS是一种系综MD方法,这意味着对完全相同的分子体系进行多个独立的MD模拟以增强模拟及其结果的统计稳健性。同样地,我们可以考虑以系综的方式运行REINVENT。类似于MD,RL方法本质上是高度随机性的,因此建议也执行多个RL计算。有多种选择可以这样做。我们可以在一个ChemProp模型上启动多个REINVENT计算。我们也可以利用通过多折交叉验证生成的多个ChemProp模型,因为每个折中还可以提供几个系综32,正如我们在本文中所做的那样。然而,需要注意的是,查询多个这样的模型会增加推理时间;实际上,缩放规模为O(N),其中N是模型的数量。在这里,单个推理步骤的时间成本约为1秒,并且进行了300-500个RL步骤,那么25个模型将需要大约一天的时间(基于本研究中的硬件)。因此,从各个模型并行推理并在事后计算统计数据将会更加高效。这样,我们也可以将其用作不确定性量化(uncertainty quantification,UQ)的评估。在RBFE研究中14已经证明了这一点,但是,在获取函数中包括其不确定度测量仅对AL性能产生了微弱的影响14。或者,可以通过改变给定网络的几何形状或使用上述完全不同的模型算法来构建多个独立的替代模型(surrogate models),这可能产生不同质量/保真度的模型,或者采用多保真度(multifidelity)方式6,22,23,其中异质数据用于创建单一的替代模型(surrogate models)。
当使用大量或容易获得的数据(对接,BindingDB数据)进行训练预训练代理模型(surrogate model),并且将昂贵的高保真数据(ABFE)以小批量添加到代理中时,人们已经证明多保真度方法特别有效23。然而,作者指出,这种方法可能被仅基于高保真度数据构建的代理模型所超越。在该研究中,代理模型基于10,000-13,000个数据点。值得注意的是,他们的生成模型88似乎是在MOSES基准化合物数据集89上训练的,该数据集是ZINC 12 Clean Lead数据集类先导(“rule of 3.5”)子集90的一部分。REINVENT28 prior不限于类先导化合物。在该研究中一些产生的分子的分子量超过500 Da。我们还注意到,他们的生成模型需要QED的权重比ABFE组件的高出两倍,这表明该模型难以产生高质量的类药化合物,也可能降低采样效率。我们的ABFE组件与QED组件权重比例为3:1。
5. 结论
本文介绍了一种用于从头设计蛋白质配体的主动学习协议,其中生成式AI REINVENT与使用系综MD模拟和MMPBSA基于物理的ABFE配体打分相结合。其中MMPBSA使用ESMACS模拟协议的粗粒度版本。该协议称为生成式主动学习(generative active learning,GAL),应用于两个测试用靶标蛋白:3CLpro和TNKS2。在两个案例中,都产生了具有大结合亲合力且结构多样、骨架有显著差异的化合物。同时,发现这些结构与最初用于训练的GAL工作流代理模型的结构非常不同,并有效地探索了化学空间的不同区域。我们发现了许多化合物,在3CLpro的案例里,配体的结合亲合力超过广泛使用的代理对接模型中产生的结构;在TNKS2案例里,那些配体已被实验确认对TNKS2表现出强结合亲合力。
TNKS2的狭窄结合口袋导致了配体明确的结合模式,这提高了GAL替代模型结合亲和力的精确度,从而在较少的GAL迭代中能够找到具有优化的结合亲和力结构。相比之下,3CLpro的特点是一个较大的开放结合口袋,允许结合模式的变化,并因此产生了精度较低的替代模型,需要更多的GAL迭代。然而,在这两种情况下,GAL都取得了成功:对于TNKS2来说,只需要3-4个迭代步骤就可以使生成的结构在化学空间内收敛;而对于3CLpro,则需要5-7个迭代步骤。这种GAL的收敛可以描述为从第一个迭代步骤中的化学空间探索到更有利用性的阶段,即在最后几个迭代步骤中发现并优化结构的过程。这个转变并没有被明确控制,所有的GAL步骤都是使用相同的自动化协议进行的,包括获取函数。
测试了不同GAL训练批量(batch sizes),得到了定性相似的结果。然而,在TNKS2案例里,较大的批量导致更精确的替代模型,以额外的计算成本加速了GAL的收敛。当比较不同训练量的计算效率时,发现较小的批量在大多数情况下更为有效。考虑到GAL对所有测试批量都成功执行,应根据计算效率选择合适的训练批量,本研究中偏向于100-250个分子的批量。
总的来说,尽管显然还有进一步改进的潜力,但我们的REINVENT-ESMACS GAL协议在当前形式下已成功应用于两个非常不同的靶蛋白作为新化合物想法的生成器。我们已经证明了该协议,在整合到更大的工作流程中评估其他重要化合物属性并进一步优化精选化合物时,能够进行从头药物设计以缩短药物发现中的设计-制造-测试-分析周期。这证明了,当使用系综模拟可靠地控制不确定性时,主动学习和基于物理的方法联合使用具有巨大潜力,并且这在大型超级计算机上是高效的。
文献
略,请查阅原文。
联系我们
想在自己的项目中亲自使用REINVENT调用OEDocking/FRED进行主动学习计算,请联系我们获取免费的试用版;你还可以采购软件或委托研究与我们进行项目合作。
- 电邮:info@molcalx.com
- 电话:020-38261356
原创文章,作者:小墨,如若转载,请注明出处:《REINVENT与精确的结合自由能排序相结合的生成式主动学习》http://blog.molcalx.com.cn/2025/02/01/gen-ai-active-learning.html


