
摘要:ROS1重排占非小细胞肺癌患者的1-2%,然而在临床上还没有选择性靶向ROS1的疗法。之前对具有选择性的强效ROS1抑制剂(相对于选定的反靶点TrkA)的认识,使得虚拟筛选成为这个项目中发现苗头化合物的方法。基于配体的虚拟筛选专注于识别与已知活性化合物具有相似3D形状和药效团的分子。为此,我们转向了阿斯利康的虚拟库,估计涵盖了可合成的按需制造的1015个分子。我们使用云计算支持的FastROCS技术搜索了整个虚拟空间中的1010化合物子集。基于化合物特性和药物化学评估,优先选择了少量特定库,并用可用的砌块进一步枚举。经过对ROS1结构的对接评估后,合成最有希望的苗头化合物并进行了生物活性测试,结果发现了几个强效且选择性的系列化合物。其中最好的ROS1抑制剂达到nM级别,对TrkA的选择性超过1000倍,并且根据初步建立的SAR,这些化合物有望可被进一步优化。我们的前瞻性研究描述了概念上简单的形状匹配方法如何通过搜索超大型虚拟库来识别强效且选择性的化合物,证明了此类工作流程在早期药物发现中的适用性和重要性。
原文:Petrović, D. et al. (2022) “Virtual Screening in the Cloud Identifies Potent and Selective ROS1 Kinase Inhibitors,” Journal of Chemical Information and Modeling, 62(16), pp. 3832–3843. Available at: https://doi.org/10.1021/acs.jcim.2c00644.
编译:肖高铿
前言
ROS1基因重排导致完好无损的ROS1酪氨酸激酶结构域与另一条染色体的伴侣蛋白之间发生融合1。这些情况占非小细胞肺癌患者的约1-2%,然而,目前临床上还没有专门设计的选择性靶向ROS1治疗药。实际上,ROS1抑制剂的临床潜力已经通过使用诸如lorlatinib2(一种ALK大环抑制剂,同时也抑制ROS1)和entrectinib3(一种泛Pan-TRK激酶抑制剂,同样作用于ALK和ROS1)之类的药物得到了证明。我们研究前提假设是,专一的、选择性的ROS1抑制剂可能为患者提供更好的疗效和/或耐受性。为此,我们将TrkA作为一个密切相关但希望避免作用的反靶标(antitarget),以推动我们实现选择性。基于TrkA与ROS1在结构上的同源性,及其药理学和安全性效应可能潜在地影响我们所识别的任何ROS1抑制剂的后续评估,我们选择TrkA作为激酶组的替代靶标进行选择性评估。
虚拟筛选(VS)是一种流行的计算苗头化合物发现方法,近年来,该领域见证了显著的方法发展,这些发展通常围绕机器学习算法和广泛的基于物理的建模。此外,这一领域正从搜索较小的、现成的数百万分子库转向搜索大几个数量级的按需定制虚拟库7-9,例如Enamine REAL10,11、WuXi GalaXi12或Mcule Ultimate13,以及辉瑞14或礼来15等公司的专有数据库。通常,搜索包含数十亿个虚拟分子的枚举化学空间需要大量的硬件资源,这通常是本地集群难以达到的。相反,可以通过使用云计算基础设施对计算进行大规模并行处理16。
基于结构的虚拟筛选在已发表的文献中占有重要地位。例如,已有报道通过暴力对接数亿个化合物来识别针对多个药物靶点的有效苗头化合物17-19。大规模对接计算的效率可以通过深度学习方法20或基于合成子的评估21(注:这里的“基于合成子的评估”指的是将分子分解为更小片段的合成子,然后根据这些片段预测整个分子的性质)来提高,以实现对数十亿个化合物的筛选。此外,自由能微扰(FEP)方法可以被整合到虚拟筛选流程中以显著提高命中率22,而主动学习FEP方法的应用可以节省大量的计算资源23。尽管依赖于对接的方法在识别新化学类型方面非常有效,但是盲对接计算不仅耗时而且常常假阳性率较高。基于配体的方法依赖于估计数据库中的化合物与已知活性化合物之间的分子相似性,并且通常优于对接算法24,25。特别是,基于形状的虚拟筛选方法可用于识别与参比分子具有高度3D相似性的新化学类型结构26-29。鉴于形状匹配计算的出色速度,这些技术可以在设计-制造-测试-分析(DMTA)工作流中得以实施,用于苗头化合物优化的迭代。因此,结合基于配体和基于结构的方法可以提升虚拟筛选的性能表现30-32。
在本研究中,我们描述了基于配体和基于结构的虚拟筛选工作流在识别强效且选择性的ROS1抑制剂方面的前瞻性应用。作为主要的基于配体的方法,我们依赖于一组已验证的ROS1抑制剂,这些抑制剂大多显示出对TrkA的选择性,我们的目标是识别新的苗头化合物系列。为此,我们使用之前发表的基于FastROCS的工作流16,在云端数百张GPU上并行计算,搜索了阿斯利康虚拟库中1010化合物的子集。进一步对识别出的最有前景的骨架进行枚举,以访问完整1015分子虚拟空间中可用的化合物。初步的苗头化合物进一步通过对接进行分析,合成了其中两个系列化合物并测试其对ROS1和TrkA的抑制作用。这一应用展示了概念上简单的形状匹配方法如何用于从超大型化学库中识别新型、强效且选择性的抑制剂。
结果与讨论
ROCS查询式的开发(Query Development)
选择了五个分子作为ROCS虚拟筛选的查询式(query)。化合物1-5(方案1)通过多种不同的方法被识别出来,包括挖掘激酶组筛选数据、分析现有激酶项目高通量筛选(HTS)结果以及一个内部的激酶AI模型。该AI模型经过训练,能够识别出相对于TrkA具有选择性的化合物。每一种方法重点都是寻找那些对ROS1表现出优异活性并且相对于TrkA有强大选择性优势的先导化合物。
方案1. 对TrkA具有稳健选择性优势、强效的ROS1抑制剂分子结构:IGF-1R酪氨酸激酶抑制剂33(1),Crizotinib衍生的ALK抑制剂34(2),Temuterkib35(3),Lazertinib36(4),以及Lorlatinib2(5)。
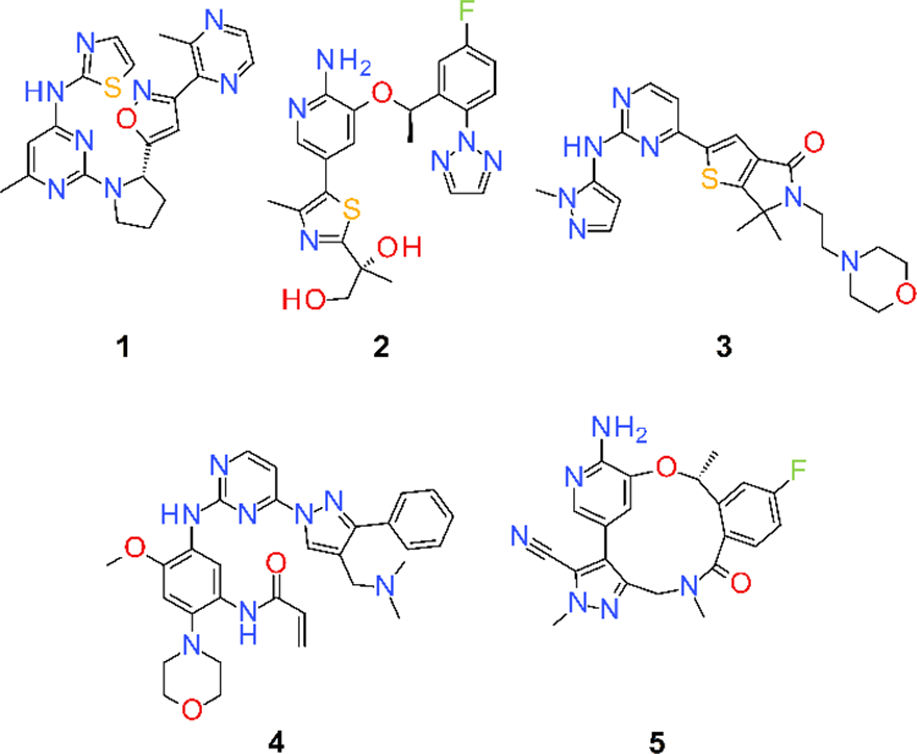
我们解释了化合物1、2与ROS1的共晶结构(PDB ID:7Z5W和7Z5X,表S1),而化合物5的结构是现成的(PDB ID:4UXL)。剩余查询式分子的生物活性构象是通过对接到ROS1来预测的,并用于指导基于配体的虚拟筛选。本质上,ROCS之类的技术根据分子的形状相似性和颜色特征(即药效团)的分布将分子在三维空间中进行叠合25,27。查寻分子的形状和ROCS-颜色特征是自动分配的(图S1)。
筛选阿斯利康内部化合物库
在阿斯利康,我们常规地依赖FastROCS38技术来搜索公司现有的分子库,不仅在开会时用来设计、快速构思创意,还作为日常更全面的虚拟筛选方法。在一张GPU上,搜索106个分子的空间大约每条查询式仅需要1分钟的时间,这对于验证开发的查询式并选择一小部分化合物进行生物测试来说已经足够快了。在搜索过程中,化合物数据库中每个构象的形状会被逐个叠合到查询式的3D形状上,并通过刚性优化过程对齐以使得体积重叠最大化。然后可以通过形状和颜色特征(即药效团)的Tanimoto分数来表示两个分子之间的相似性,每个分数的取值范围在0到1之间。它们的总和记为Tanimoto combo(Tc)打分,该分数的范围是从0到2,数值越高表示化合物越相似。
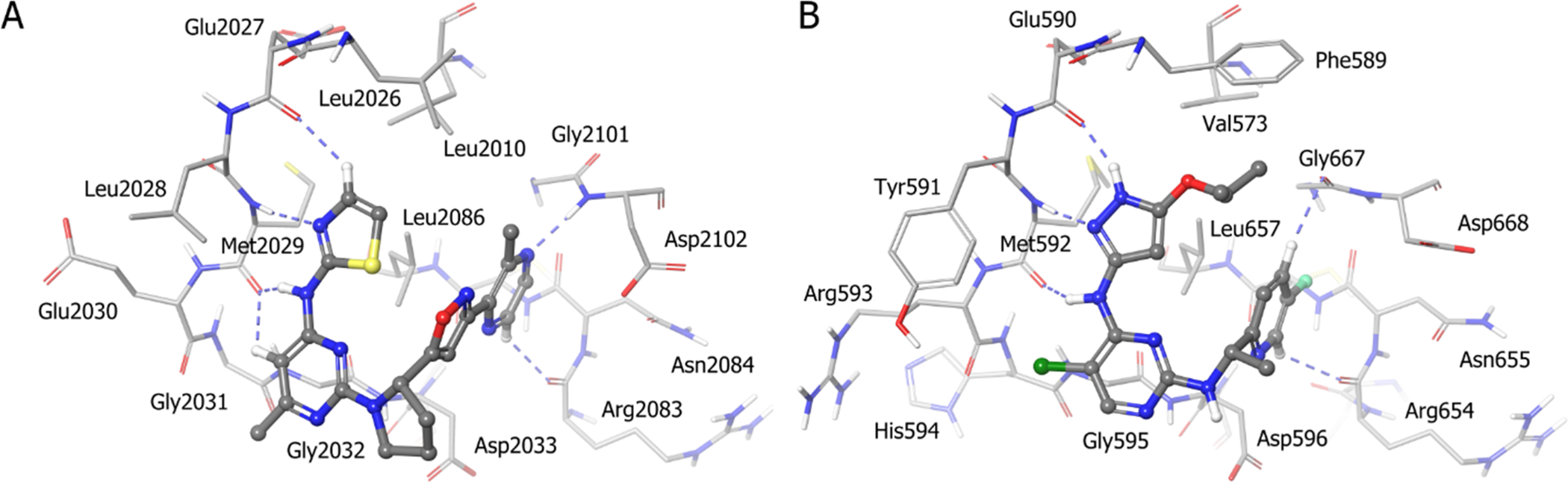
图1. (A)ROS1与化合物1的共晶结构(PDB ID: 7Z5W)(B)4-胺基吡唑并嘧啶抑制剂AZ-234−64-6与TrkA共晶结构(PDB ID: 4AOJ)
在本研究中,FastROCS使用全部的五个查询式筛选公司内部数据库中的分子,识别出2900个\(Tc\ score ≥ 1.2\)的苗头化合物。由于FastROCS是基于总体相似性给化合物打分的,因此有可能一个分子虽然获得了良好的颜色Tanimoto得分,但却缺乏关键的药效团特征。我们去除那些缺乏ATP结合位点主要药效团特征(即与激酶铰链区结合的供体-受体-供体模式,见图1A)的化合物以及已知的ROS1化学类型的化合物,剩下的1700个化合物使用Glide SP协议对接39,40到ROS1受体网格(PDB ID:3ZBF和4UXL)。对于每个化合物,基于对接分数选择最佳受体。对对接打分与已知活性分子相当或更好的苗头化合物进行视觉检查,我们选择了400个苗头化合物进行生物活性测试的。由于我们的假设是从已经具有选择性的抑制剂出发进行形状搜索可以直接识别其他有选择性的苗头化合物,因此没有对接到反靶点上。通过ECFP6指纹进行相似性聚类分析,发现所选分子具有结构多样性,共54个簇与16个单例。
采用ADP Glo检测法,通过测量由ROS1/TrkA催化的底物肽ATP依赖性磷酸化产生的ADP量,来评估分子是否能阻止这一专一的磷酸化事件的发生。生物筛选证实,有280个化合物对ROS1表现出\(IC_{50} <10 μM\)的活性,命中率为70%。在这些化合物中,有80多个苗头化合物显示了对TrkA有10多倍的选择性窗口,而至少有8个苗头化合物被确定具有不低于100倍的选择性(图2)。具体来说,我们不仅识别出了新的ROS1活性化合物,而且还保持了对关键反靶点(antitarget)的选择性,这证明了这种虚拟筛选工作流的强大苗头化合物发现能力。
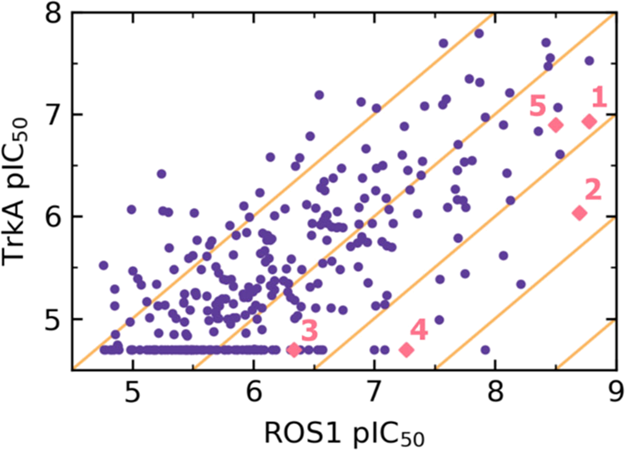
图2. FastROCS虚拟筛选阿斯利康的企业库识别出的400个潜在的ROS1抑制剂重新用Glide SP进行打分。在确认的280个苗头化合物中,有8个亚μM级别的ROS1抑制剂对TrkA显示出100多倍的选择性。在高达20μM浓度下对TrkA不能监测到活性的化合物,其pIC50值绘于4.7处。查询式分子1-5的活性以粉红色菱形表示。
其中13个化合物的ROS1 IC50值在个位数字nM水平,其中活性最强抑制剂的IC50=2 nM。然而,大多数化合物显示出较差的选择性特征,其TrkA IC50值在亚μM级别。与此相反,化合物6被鉴定为一种ROS1 IC50 = 13 nM的抑制剂,并且对TrkA具有3900倍的选择性窗口。这一苗头化合物是作为较大簇的一部分(表1)从查询式3中选出的。此外,这些化合物的特点是具有一个新型的ROS1头部基团,最初用在细胞外信号调节激酶(ERK)项目中42,43。
表1. 通过公司内部化合物库虚拟筛选识别出的ERK头部基团系列的构效关系
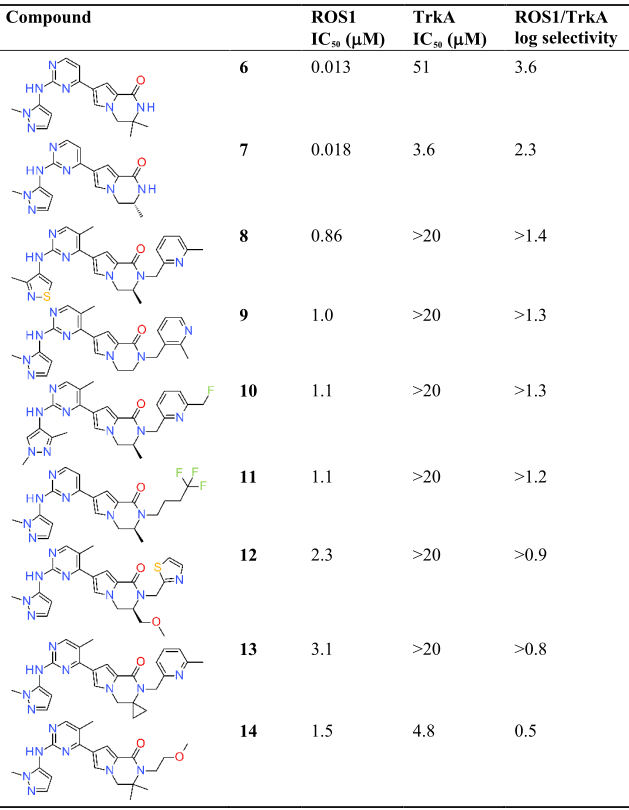
化合物6,含有二甲基取代的双环内酰胺,是识别出的活性最高和选择性最大的化合物。化合物7去除了6中偕甲基的一个甲基,保持了活性但选择性有所下降,这表明这是一个值得进行构效关系(SAR)研究的区域。苗头化合物8和9对ROS1具有亚微摩尔活性,但在TrkA上没有活性。这一对结果表明,在嘧啶上的甲基取代是可以接受的,内酰胺的NH可以被杂苄基取代,并且内酰胺上的甲基和去甲基化修饰都能提供有效的抑制剂。此外,化合物8证明吡唑可以被噻唑替代,尽管在没有专门匹配分子对的情况下很难确定每个结构变化的具体贡献。化合物10-13对ROS1都具有低微摩尔活性,但在TrkA上没有活性。它们提供了更多关于耐受性的SAR信息,包括对于吡唑(+甲基,10)、其他杂环(10, 12)和烷基(11)取代以及内酰胺的醚(12)和环丙基(13)修饰的耐受性。最后,具有N-烷基取代的化合物14在这一系列中表现出最差的选择性。
阿斯利康虚拟库
除了搜索较小的、现成的化学数据库,比如我们的公司库(包含106个分子)之外,虚拟筛选使我们能够扩展到那些使用化学信息学方法枚举出的、尚未合成的虚拟化合物库。搜寻更多化合物背后的理由是能够识别到打分更高的苗头化合物,并为先导化合物的优化提供更高质量的起点。实际上,这类虚拟库的大小和组成受限于筛选技术的吞吐量,以及快速合成识别出的苗头化合物的能力。随着云计算的最新进展,诸如FastROCS之类基于形状的方法可以扩大至数十或数百个GPU,提供足够的计算能力来筛选比现成化合物库大几个数量级的虚拟数据库。
路线2. 阿斯利康虚拟库的枚举合成路线
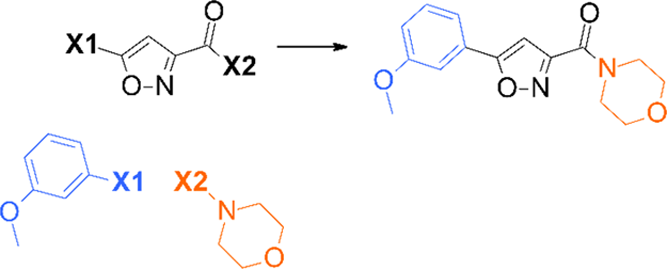
a. 骨架(黑色)编码有1到4个连接点,在这种情况下,准备好的砌块(蓝色和橙色)直接在X1和X2点处通过Suzuki偶联反应和酰胺偶联反应分别连接上去。
阿斯利康虚拟库(AZVL,AstraZeneca Virtual Library)的开发之前已有描述16。简而言之,化学空间基于4000多个母核或“骨架”构建,每个骨架都有既定的合成方案44,并配以适当类别的试剂。如方案2所示,示例骨架有两个连接点,其中X1位置通过与一组硼酸砌块进行Suzuki反应连接,而X2位置则使用一系列胺砌块进行酰胺偶联反应。例如,1300种硼酸和3500种胺可以生成包含460万个分子的虚拟库。在这个协议下,利用所有可用的砌块对所有骨架进行完整或部分枚举,便形成了虚拟库,由于具有既定的合成方案和现成的砌块,成功合成化合物的概率得到了提高。
AZVL骨架具有1~4个连接点,根据砌块的可用性,我们估计完全枚举的虚拟空间将包含1015个分子。我们之前的研究表明,随机选取的1010个分子的子集具有足够的多样性,并且能够很好地代表整个库,同时使用现有的云计算基础设施可以对其进行高效搜索。
在云上进行高通量虚拟筛选
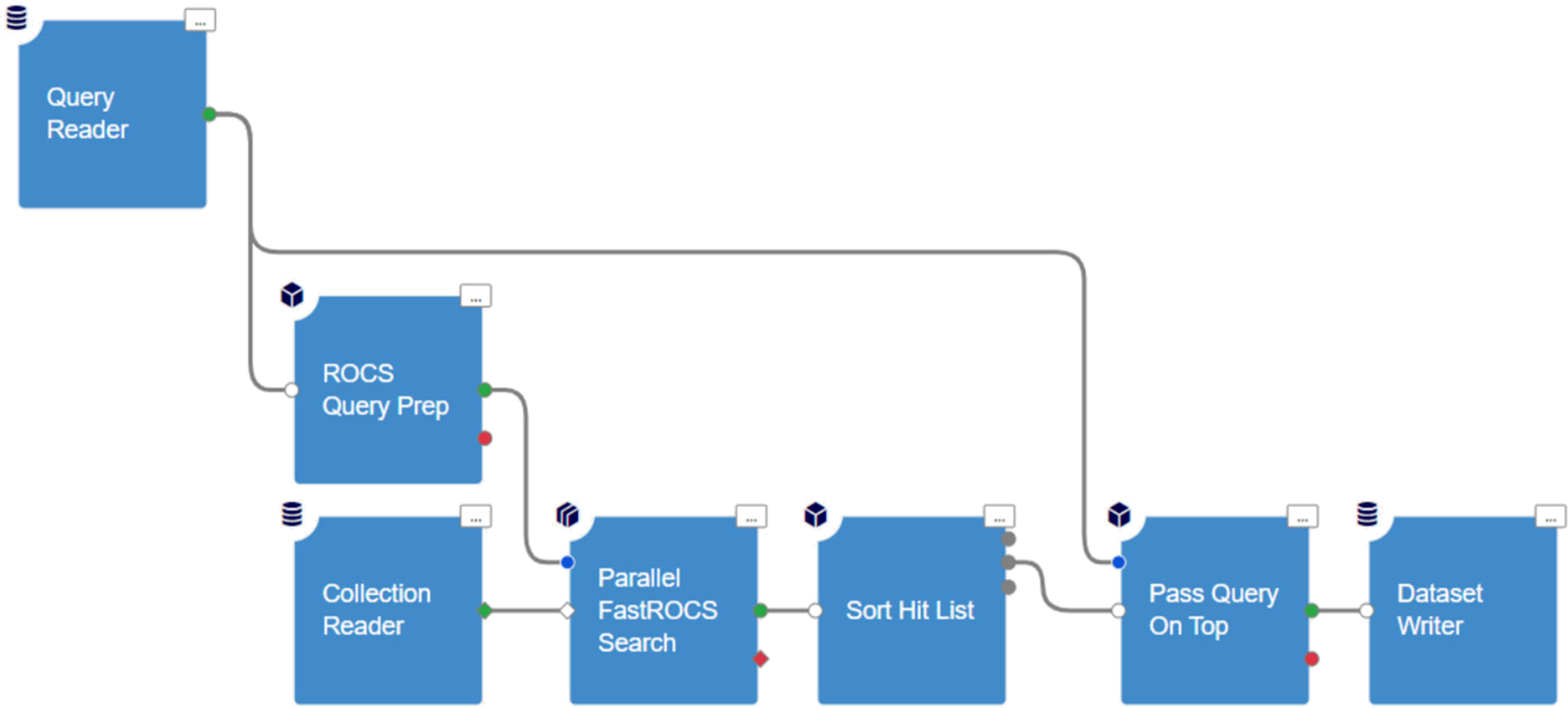
图3. Orion的FastROCS虚拟筛选工作流
搜索1010个AZVL分子的能力是由FastROCS工作流实现的,如图3所示,该工作流部署在我们的Orion45,46云计算平台上。简而言之,此工作流需要两个输入:查询式读取器(Query Reader)模块处理查询式的3D结构,而数据库读取器(Collection Reader)模块加载OMEGA准备好的虚拟库并分发。查询式记录通过ROCS Query Prep模块处理,在此过程中为从输入端口(intake port)读入的分子添加氢原子和颜色原子,为FastROCS计算做准备。修改后的分子通过成功端口(success port)发出,并用于初始化并行FastROCS搜索(Parallel FastROCS Search )模块——这是工作流的核心部分——对查询分子与AZVL中所有构象进行快速形状和颜色比较,根据定义的相似性打分(通常是Tanimoto combo)对其进行排序。此模块运行于GPU上,通常使用Amazon Web Services (AWS)的g3.4xlarge(120 GB RAM)基础设施47,并且可以根据资源可用性扩展到数百个实例同时运行。打分靠前的苗头化合物作为记录通过成功端口发出并读入苗头化合物排序列表(Sort Hit List)模块,该模块通过基于指定相似性打分的浮点值对结果进行排序来积累一个给定大小的苗头化合物列表。最终的苗头化合物列表被发送到hit_list端口,其余记录则被丢弃。Pass Query On Top模块将查询式的3D构象添加到苗头化合物列表输出的开头,以便于视觉检查,最后数据集写模块(Dataset Writer)将记录输出到一个新的Orion数据集中。针对一个查询式搜索整个AZVL平均需要一个小时,工作流可扩展到200多个GPU。从AWS临时租用硬件进行此类计算的相关成本通常不到100美元。
在初步的虚拟筛选(VS)中,我们对ROCS查询式1-5进行了FastROCS搜索,将每个查询式排名前1万的苗头化合物(最高的Tc打分)写出。Tc打分分布如图4A所示,结果表明查询式1、3和5检索到的苗头化合物的Tc打分均\(≥1.2\),而对于查询式2和4,则分别仅识别出222和450个打分高的苗头化合物。通常,每个查询式可识别出60到270个不同的骨架,其中大约一半的骨架苗头化合物是单例(指的是只包含一个化合物的簇)或不超过10个化合物的小簇(图4B)。我们主要关注那些具有较高打分苗头化合物的较大簇。检查含有超过10个苗头化合物的簇,并且至少有一个苗头化合物的Tc得分\(≥1.2\),每个查询式可识别出15到60个骨架(图4C)。由于一些骨架在多次FastROCS搜索中被独立识别出(图S2),最终不重复骨架的列表包括了162个不同的库。
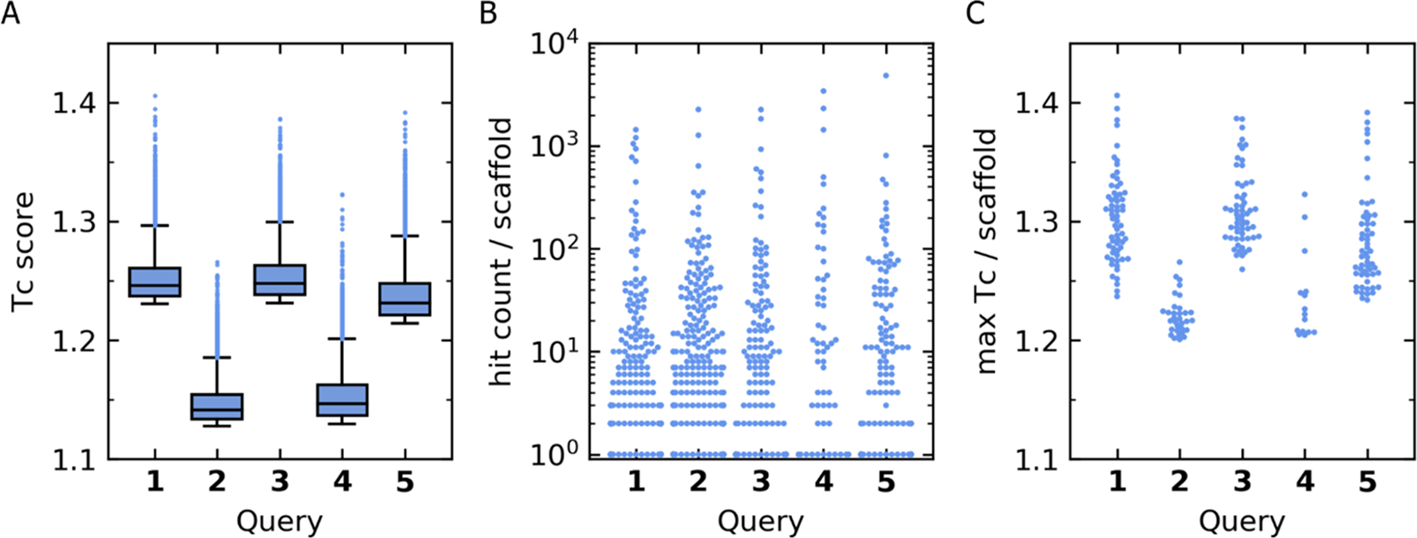
图4. FastROCS使用查询式1-5对AZVL进行虚拟筛选。(A)每个查询式检索到的前1万个苗头化合物的Tc打分分布。(B)所有苗头化合物的骨架分析。每个数据点代表一个独特的骨架,y轴显示了每个骨架中识别出的苗头化合物数量。一些骨架被多个查询式独立识别到。(C)对所有含有10个以上苗头化合物且至少有一个化合物的\(Tc\ score≥1.2\)的骨架进行分析。每个数据点代表一个独特的骨架,y轴显示了在骨架簇中识别出的最大Tc打分。
如前所述,一些FastROCS苗头化合物可能缺乏能够与ATP结合位点发生相互作用的关键化学特征,因此这些化合物被去除掉。下一步,从所有库中选择代表性苗头化合物对接到多个受体(3ZBF、4UXL、7Z5X),并识别出得分较高的化合物。基于各系列内的理化性质预测、结构新颖性、与项目中其他资产的差异性、估计的合成化学资源需求以及常规的药物化学评估,有7个骨架被进一步推进进行库枚举,其中一些是由多个查询式识别出来的,详见表S2总结。
尽管由于技术限制并未对完整的1015个AZVL分子进行全面搜索,但一旦从小规模的初步虚拟筛选(VS)中优先选出少量骨架,就可以围绕这些骨架进行完整库枚举,以在二次VS期间进一步富集苗头化合物。图5总结了这一两步协议,使我们能够在对一个显著较小的子集进行暴力搜索的同时,从整个1015个AZVL分子中找到并恢复所有令人感兴趣的分子。在当前情况下,对所有七个骨架进行完整枚举将产生近1700万个虚拟分子。虽然使用FastROCS搜索这个空间是可行的,但我们基于砌块分析减少了复杂性。具体来说,在大多数库中,大量砌块要么太大而无法适应特定的蛋白质口袋(基于初次VS中的对接分析),要么缺乏所需的激酶铰链结合药效团特征。采用基于知识的方法预先过滤砌块库,并仅枚举具有高成功概率的化合物,这导致在所有七个库中总共约有4.5万个虚拟化合物。经过性质过滤去除Lipinski空间之外的、不具吸引力的化合物,并通过对接ROS1和视觉检查后,我们决定合成两个库。库A包含近7000个化合物,用所有五个查询式识别,从中挑选了70个化合物进行合成。在这个过程中,2-(methylthio)-pyrimidin-4-ol通过SNAr反应在2位引入两个4-吗啉苯胺砌块之一作为溶剂口袋基团,随后进行氯化并通过与35个硼酸砌块的Suzuki反应实现多样化。库B包含2000多个化合物,由查询式1、3和4识别,其中2,4-二氯-6-甲基嘧啶在4位与噻唑-2-胺选择性地通过Buchwald-Hartwig偶联结合,然后通过与16种不同的仲胺的SNAr反应实现多样化。
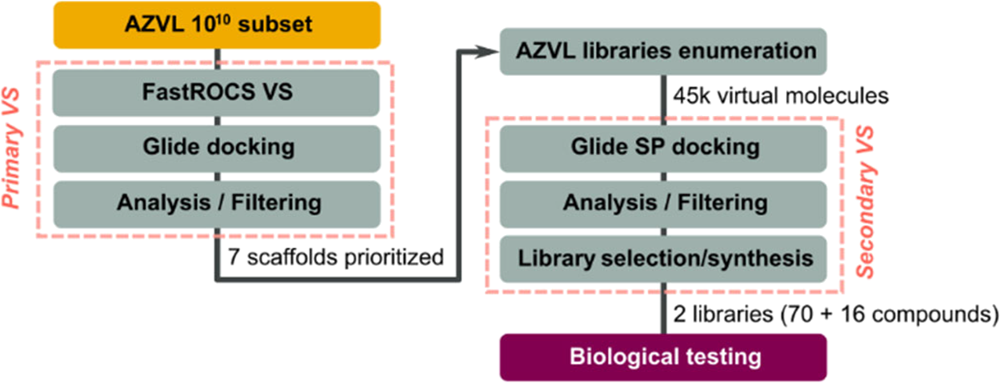
图5. 两步虚拟筛选工作流使得从含有1015化合物的AZVL中获取化合物成为可能
苗头化合物确认(Hit Confirmation)
在库A中,化合物是基于它们与lazertinib(4)的3D相似性来选择的,其特征在于具有一个带有苯基吗啉取代的2-氨基嘧啶的头部基团。通常,R1 = OMe取代的化合物相对于其R1 = H匹配分子对显示出略微更好的ROS1活性,同时对TrkA具有更高的选择性(表2)。在最有效的苗头化合物中,化合物16的IC50=13 nM的ROS1抑制剂,对TrkA的选择性接近5000倍。这个化合物携带了一个与从公司内部库中识别出的ERK样的化合物相似的头部基团。
表2. 通过对AZVL虚拟筛选识别的库A的SAR
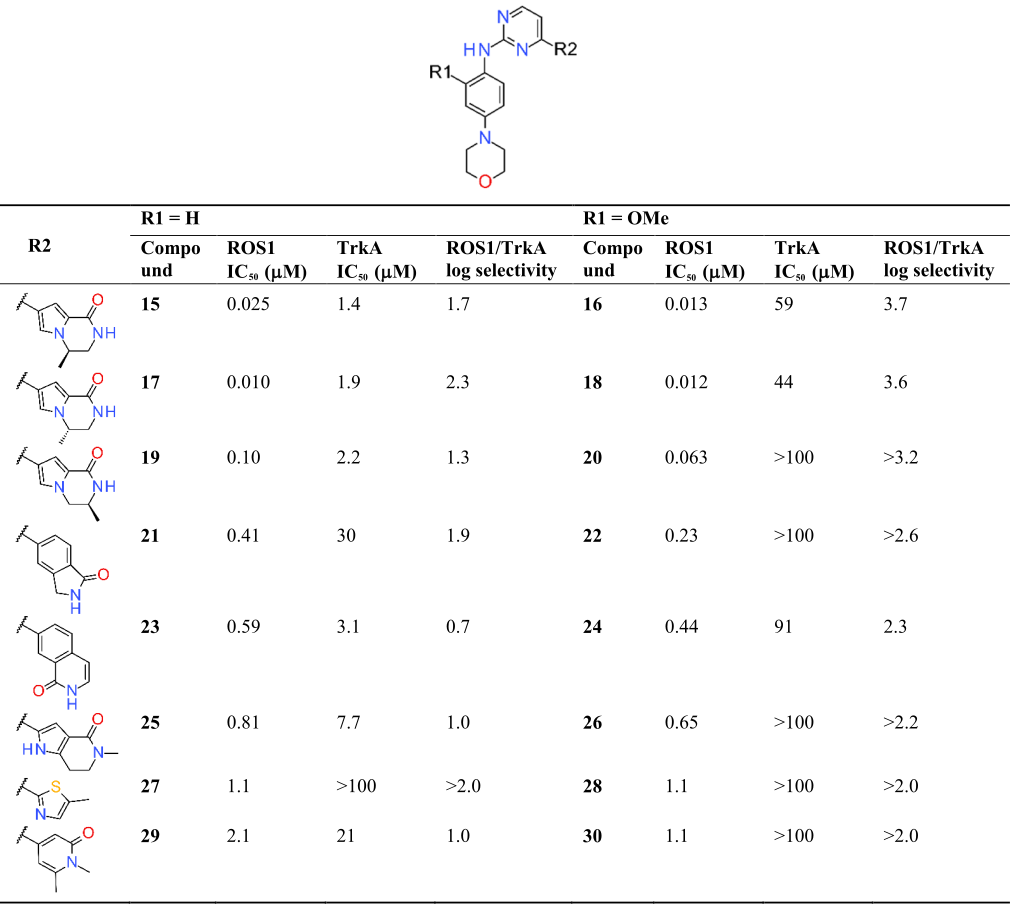
关于构效关系(SAR),化合物17和18在母核甲基取代位置具有与15和16相反的立体化学,显示出相似的活性和选择性。化合物19和20交换了甲基取代基的位置,相对于其匹配分子对17和18,显示出大约四倍的活性下降。此外,21至26展示了各种双环内酰胺,有或没有N-取代的化合物在抑制ROS1方面都达到了亚微摩尔水平。最后,化合物27至30表明双环内酰胺可以被简单的杂环如噻唑和吡啶替代,尽管这会导致活性的损失。
在库B(表3)中,也发现了具有个位数纳摩尔级别的ROS1抑制剂,如38号化合物,它们显示出令人鼓舞的选择性(\(>25\)倍)。最令人感兴趣的苗头化合物是31号化合物,它是一个IC50=97 nM的ROS1抑制剂,对TrkA的选择性窗口超过1000倍。该化合物携带了一个新颖的2-氯-4-(甲氨基甲基)苯腈基团,这为先导优化提供了一个有前景的起点,具有提高活性和调节类药性质的潜力。
表3. 通过对AZVL虚拟筛选识别的库B的SAR
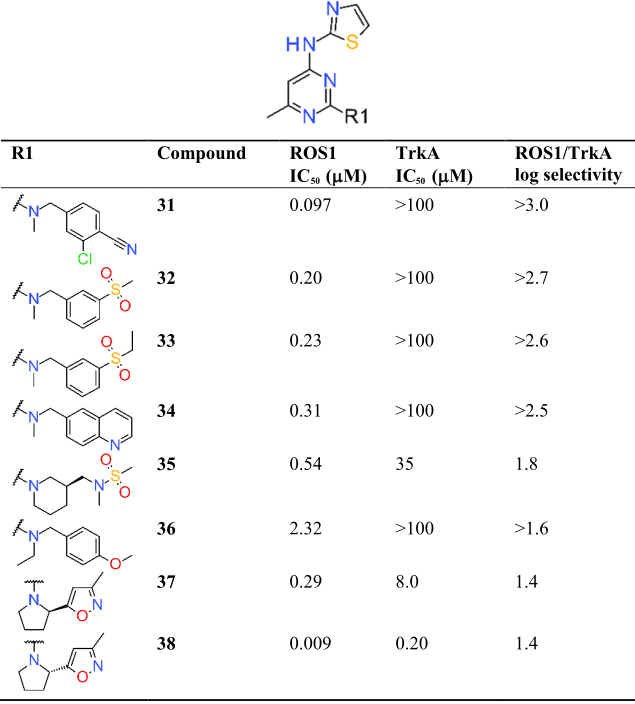
化合物32和33表明,仅在间-位引入一个极性的砜只会轻微降低活性,并且仍能保持对TrkA的\(>100\)倍选择性。苗头化合物34显示其喹啉基团也被很好地耐受,并且没有显示出可辨别的TrkA活性,而35则表明从N-烷基取代基用带有磺酰胺基团的哌啶环化仍然具活性,其中磺酰胺基团的位置与上述砜类似。化合物36有一个延长的N-乙基取代基和一个甲氧基芳基取代基,尽管失去了一些活性,但对TrkA仍保持了极好的选择性。最后,37和38展示了化合物1中存在的噁唑-吡咯烷取代基的截短版本。这些立体异构体在与ROS1结合时表现出\(>30\)倍的选择性偏好,其中38是从这组中识别出的活性最高的化合物(IC50 = 9 nM)。有趣的是,这种选择性偏好在其与TrkA的结合中也完全一致,从而在这两种情况下都保持了相同的选择性差距。
我们寻求从激酶组数据以了解化合物31与原始虚拟筛选先导物1相比的激酶谱特性。如图6所示,化合物1显示出较为广谱的抑制,在1 μM浓度下对17种激酶的抑制率超过了80%。相比之下,化合物31的表现显著不同,在1 μM浓度下仅对ROS1的抑制率超过了80%。值得注意的是,31在1 μM浓度下对任何其他激酶的抑制率均未超过50%,这使其成为一个针对ROS1优化真正选择性的起点。
最后,选定化合物的生物活性、理化性质和代谢稳定性数据如表4所示,允许比较新型虚拟筛选苗头化合物6、16和31与其起始点分(分别为3、4和1)的性质。对于化合物6,通过降低亲脂性实现了活性的提高,从而改善了亲脂配体效率(LLE),并且与化合物3相比,在人体肝微粒体中的转化率更低。对于化合物16,尽管活性提升较为有限,但同样通过降低亲脂性实现,并因此大幅提升了LLE。这导致其溶解性增加,并且在人肝微粒体和大鼠肝细胞中的转化率都低于化合物4。最后,相较于化合物1而言,化合物31显示出活性有所下降,且由于相对较高的亲脂性,其性质表现较差。然而值得注意的是,它在整个激酶组中对ROS1表现出极高的选择性,这一点为未来的优化提供了令人感兴趣的起点。
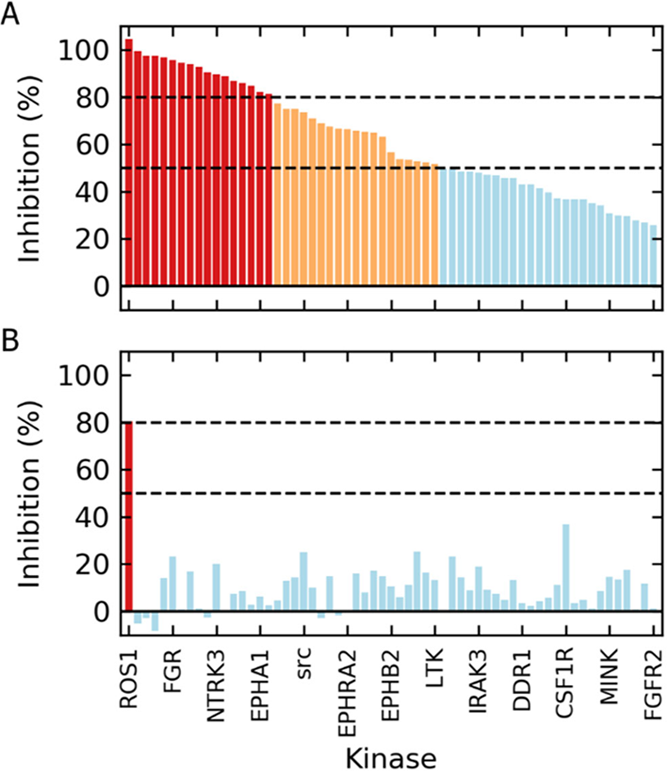
图6. 化合物在1 μM浓度下对400多种激酶的选择性(表格化数据见表S3):(A) 1和(B) 31。
结论
在本次筛选中,虚拟筛选识别出了三个不同系列的高效且选择性的ROS1抑制剂(表4)。首先,通过使用查询式3筛选阿斯利康的公司库,我们从ERK项目中识别出了一种新的头部基团,如化合物6所示。这个ERK头部基团将ROS1的活性提高了40倍,并显著改善了LLE(亲脂配体效率)。此苗头化合物还显示出ROS1(IC50 =13 nM)与TrkA(IC50 =51 μM)之间极高的选择性(\(>1000\)倍)。剩下的两个系列是通过多个查询式在云端进行的大规模虚拟筛选从AZVL中被识别出来的。库A包含了与lazertinib(4)相似的溶剂口袋基团,其中16号化合物携带了一个与ERK系列相关的头部基团。相比4号化合物,16号化合物对ROS1的活性提高了4倍,同时显著提升了LLE并保持了相似的选择性特征。最后,库B围绕查询式1中的氨基噻唑头部基团展开,识别出了一种新型的溶剂口袋基团,以31号化合物为例。尽管这个苗头化合物在ROS1活性和LLE方面相比查询式有所下降,但它在TrkA和泛激酶选择性方面有了显著改进。总之,这些系列为开发特意设计的选择性ROS1抑制剂的从苗头到先导化合物优化阶段提供了有希望的起点。
虚拟筛选是阿斯利康常用的苗头化合物发现方法。除了搜索我们的企业库和商业数据库外,我们还投入大量精力搜索如AZVL这样的虚拟库。这得益于在Orion云计算环境中实现的FastROCS技术,可以高效地浏览数十亿个分子。我们发现,结合这种简单的基于形状的方法从超大型化学库中检索初步的苗头化合物,并使用更复杂的基于结构的方法进行苗头化合物处理,是快速推进项目的关键。因此,我们依赖于易于合成且具有高合成成功率的虚拟库分子,以便对项目的化学研究早日产生影响。在此,我们证明了相似的形状和药效团如何导致相关的生物反应,甚至在靶标活性之外的其它性质也是如此。通过从活性和选择性的ROS1抑制剂出发,我们识别出了全新系列的苗头化合物,在提升ROS1活性的同时,也改善了对TrkA和整个激酶组的选择性。
实验部分
计算
企业库的106个小分子通过Flipper50处理,以枚举未定义的立体中心和双键。对于含有超过两个未定义立体中心和双键的分子,随机选择两个进行完全枚举,其余的则固定为单一随机立体异构体。准备好的SMILES字符串数据库使用OMEGA50,51进行处理,每分子生成20个构象。3D扩展数据库在本地GPU节点上使用FastROCS服务器进行搜索,根据Tanimoto combo 打分量化最佳形状重叠对苗头化合物进行排序。在AZVL的1010子集中,每个分子包含10个由OMEGA生成的构象。此数据库使用部署在OpenEye Orion云计算平台上的FastROCS工作流进行搜索,并在200多个GPU上并行计算。苗头化合物按照最高的Tanimoto combo打分进行排序。
对接模拟使用Schrodinger Suite(v. 2019-2)。受体结构使用Protein Preparation Wizard 从PDB ID:3ZBF、4UXL和7Z5X开始准备,并使用Receptor Grid Generation工具生成对接用的网格,围绕共晶配体定义受体位点。在对接之前,小分子使用LigPrep进行准备,然后使用Glide SP算法对接到多个受体上。
报道的苗头化合物使用本地PipelinePilot节点中实现的PAINS过滤器54,55进行评估。这些化合物没有显示出任何警告,除了表2中报告的未取代的4-苯基吗啉系列(R1 = H),该系列被标记为anil_di_alk_A(478)。由于与-OMe取代的匹配分子对非常相似且通常具有表现良好的构效关系(SAR),这并未引起我们的重大关注。
化学
略
生物
略
数据与软件的获取
阿斯利康的企业库和虚拟库是专有的化学空间。用于搜索企业库的FastROCS服务器基于OpenEye Toolkits。为了搜索虚拟库,我们在Orion云计算平台上使用了基于OpenEye Toolkits的FastROCS工作流。虚拟库枚举的方法和FastROCS工作流的技术实现在参考文献16中有详细描述。所有OpenEye软件均根据商业许可证访问。对接使用的是根据商业许可证获得的Schrödinger套件。
文献
略
关于FastROCSTM
FastROCS是OpenEye开发的极速形状相似性搜索工具,可以为您的虚拟筛选和先导跃迁提供近乎即时的结果。FastROCS形状搜索的速度接近2D如指纹图谱方法,这种卓越的速度使您能够在几秒钟内对数百万个分子执行高度精确的三维形状相似性计算,与传统的二维方法相比具有显著的优势。
FastROCS的特性
- 获得近乎即时的结果,每秒处理数百万个构象
- 使用VIDA桌面可视化工具或Orion网页界面启动任务并查看随后的结果
- 用严格Tanimoto或Tversky度量来报告形状相似性
- 根据与查询的3D形状匹配质量返回叠合结果
- 叠合结果直观且视觉信息丰富
- 在一个步骤中将基于配体和基于结构的方法无缝地结合在一起为您服务
FastROCS你需要知道的事
FastROCS怎么用?
FastROCS既以工具包形式(FastROCS TK)提供,也通过OpenEye的基于网页的界面(Orion云计算平台)提供,还可以通过FTE服务提供。
ROCS与FastROCS有什么区别?
ROCS® 是一个基于形状打分的综合性工具,能够在1个CPU上以每秒100个分子的速度进行计算。FastROCS结合了ROCS的形状相似性先进科学与最新的GPU技术,使用FastROCS,您可以搜索数十亿化合物的虚拟库或通过形状对数百万分子进行聚类。通过FastROCS,3D形状相似性计算与2D方法一样快。
FastROCS plus有什么额外优势?
FastROCS Plus扩展了FastROCS在多个查询上的计算能力,并提供了选项可以对苗头化合物用形状、对接或两者进行重新打分。FastROCS Plus能在一个步骤中无缝地将基于配体和基于结构方法结合在一起为您服务。
FastROCS和FastROCS Plus —— 可信赖的科学、极致的速度和无限的规模!更快地找到您的下一个候选药物。
联系我们
想在自己的项目中使用OMEGA、FastROCS等OpenEye软件进行超大规模虚拟筛选,请联系我们获取免费的试用版;你还可以委托研究的方式使用该软件。
- 电邮:info@molcalx.com
- 电话:020-38261356
原创文章,作者:小墨,如若转载,请注明出处:《云上虚拟筛选发现强效、选择性ROS1抑制剂》http://blog.molcalx.com.cn/2025/02/04/fastrocs-virtual-screening-in-the-clound.html


