
Thompson sampling──高效的超大型按需合成库虚拟筛选方法
摘要:在过去五年中,虚拟筛选超大型按需合成库已成为药物发现项目中识别苗头化合物的强大工具。随着这些数据库增长到数十亿个分子,已经到了成本效益不再增长的点,无法再对每个分子进行虚拟筛选。为了解决这个问题,几个团队开发了启发式搜索方法来快速识别虚拟筛选的最佳分子。本文描述了一种主动学习的方法Thompson sampling(TS),通过在试剂空间中执行概率搜索来简化大型组合库的虚拟筛选,从而无需完全枚举整个库。TS是一种通用技术,可以应用于各种虚拟筛选模式,包括2D和3D相似性搜索、对接以及机器学习模型的应用。在一个算例中,通过评估数据集的1%,TS能够从基于对接虚拟筛选3.35亿个分子识别出的打分靠前的100个分子中的一半以上。
原文:Klarich, K. et al. (2024) “Thompson Sampling─An Efficient Method for Searching Ultralarge Synthesis on Demand Databases,” Journal of Chemical Information and Modeling, 64(4), pp. 1158–1171. Available at: https://doi.org/10.1021/acs.jcim.3c01790.
编译:肖高铿
前言
虚拟筛选(Virtual screenning)1,2在学术和制药工业药物发现中广泛地用于苗头化合物的识别(hit identification)。人们用各种计算方法来搜索廉价的、商业可供购买的化合物库,并识别优化的起点。当蛋白质结构可用时,对接3,4可以用来识别潜在的配体。当有一个或多个参比配体可用时,可以使用2D6(7)或3D8结构进行相似性搜索5,6。活性和非活性分子也可以用来训练机器学习模型9,10,以便对拟合成或采购的分子进行优先级排序。直到最近,虚拟筛选通常应用于包含数百万个分子的化合物库。在这种情况下,有可能对数据库中的每个分子进行全面评估。最近,一些公司开始供应超大型按需合成库11,12。这些库是由现有试剂的组合反应构建而成。通过将数十到数百种反应的组合,供应商可以组装出由数十亿个分子组成的化合物库。供应商通常会提供电子形式的可用分子结构,以便使用虚拟筛选软件进行搜索。相应的化合物往往每个售价在100至200美元之间。这些化合物通常会在6-8周内交付,合成成功率接近80%。表1列出了当前的一些按需合成库。正如我们所看到的,其中许多库都包含了几十亿个分子。
表1. 几个按需合成超大库
| Database | number of molecules | Reference |
|---|---|---|
| eMolecules eXplore | \(2.8 \times 10^{12}\) | https://marketing.emolecules.com/explore |
| Enamine REAL | \(3.5 \times 10^{10}\) | https://enamine.net/compound-collections/real-compounds/real-database |
| WuXi GalaXi | \(8 \times 10^9\) | https://wuxibiology.com/drug-discovery-services/hit-finding-and-screening-services/virtual-screening/ |
| Otava CHEMyria | \(1.2 \times 10^{10}\) | https://www.otavachemicals.com/products/chemriya |
除了按需商业合成的化合物库之外,几个团队还组合生成可以通过先前的化学反应进行合成的大型虚拟库。包括GSK、礼来13、默克和辉瑞在内的制药公司团队已经使用内部开发的反应和试剂库,组装了由108到1020个分子组成的虚拟库。还有一些具有类似目标的公共和开源化合物库。其中最完善的软件包之一是Varnek团队开发的SyntOn(原名SynthI)软件包14。SyntOn包含将试剂转化为合成子的工具以及将这些合成子组合成反应产物的程序。Warr在2022年的一篇论文中15,Cavasotto在2023年的论文中16对超大型化合物库的当前状态进行了很好的描述。
超大型虚拟库改变了虚拟筛选的范围。当我们处理包含数十亿个分子的库时,使用蛮力方法是不切实际的。虽然可以利用云资源可以将十亿个分子进行穷尽对接,大约需要花费12,000美元,但对于更大的库来说,这种做法在经济上并不实用。例如,对五十亿个分子进行穷尽对接的成本约为60万美元,超过了大多数机构的虚拟筛选预算。另一种缺点是,穷尽暴力对接所需的3D构象生成成本和磁盘空间需求。大多数对接和三维相似性方法都需要一组预先生成的构象。尽管这个过程只需要为一个库执行一次,但成本可能很大。根据Sivula最近的一篇论文17,对于15.6亿个分子的构象生成需要457,600个CPU小时,并且产生的压缩数据库(DB)消耗了超过10TB的磁盘空间。如果考虑增加这些库的规模一倍,计算和存储成本就会变得不太可行。
为了克服这些局限性,研究小组开发了新的方法来计算评估超大型库。最近的几篇论文描述了使用机器学习模型作为代理(surrogate)以加速对接计算的方法18-20。虽然对接计算通常需要每分子1到10秒的时间来生成结合模式并进行打分,但机器学习模型可以每秒处理数千个分子。在这一方法的一个早期例子中,Berenger21对接了10,000个分子,并使用对接结果生成了一个基于一维化学结构表示预测对接打分值的机器学习模型。这个模型是用大约20%的数据集构建的,随后被用来预测剩余80%分子的对接大分值。在Lit-PCBA数据集中15个靶点上,作者实现了机器学习的打分预测值与观察到的对接打分值之间的\(R^2>0.7\)。
上述的被动机器学习方法已经扩展到涉及多轮对接和建模迭代的主动学习(active learning,AL)方法。AL方法的初始步骤与上述方法类似。首先从大型数据库中选择一个子集并进行对接,然后将化学结构和对接的打分值来构建预测对接打分值的机器学习模型。此时,计算流程开始偏离上述的被动方法。机器学习模型会选择另一个子集,并对其进行对接。第二轮对接分子的选择可以有几种方法。在某些情况下,团队会选择(预测的)对接打分值的最高分子用于下一轮对接。在其他情况下,还可以在选择过程中使用额外的打分项,例如模型预测中的不确定性。这一轮对接的结果会被合并到之前的迭代结果中,并用来构建一个新的机器学习模型。对接、建模和选择的过程可以通过多次迭代继续下去。
大型虚拟库是由许多较小的试剂集构建而成。一个由十亿个分子组成的库可以从3个不同试剂各100个合成出来。超大型数据库对接的一种替代方法是将库还原为组成其的砌块(building block),对接这些砌块,并只详细描述最有前途的片段。V-SYNTHES22方法首先通过将大型组合库还原成骨架和单个合成子数据库,称为最小枚举库(minimal enumeration library,MEL)。每个MEL组件包括从骨架上连接到一个R-基团位置的一个单一合成子构造的片段。骨架上的剩余附着点用甲基或苯基封端基团填充。在创建MEL时,作者将REAL库的大小从110亿减小到了60万。然后将MEL中的分子对接到感兴趣的蛋白质中,并确定了最有前途的片段。随后一步,对被封端的R-基团进行迭代替换为相应完全阐述的分子中的取代基。最后,相应的完全枚举的分子进行对接,并记录了它们的打分。Sadybekov于2021年发表的一篇论文介绍了V-SYNTHES在识别ROCK1抑制剂方面中的应用。仅对接了一个包含110亿个分子的库的0.1%,作者实现了33%的命中率并发现了14个亚微摩尔配体。在2022年的论文中,Beroza23采取了一种类似的方法,但将分析限制在Enamine REAL库的一个约10亿个分子的子集,来自102个双成分的反应。V-SYNTHES和Beroza所用方法的一个潜在缺点是,对接片段可能无法重现与完整分子发现的结合模式。
在后续章节中,我们描述了Thompson sampling(TS)采样24,这是一种利用超大型库的组合性质的概率方法。TS通过在试剂空间而不是产物空间工作来实现效率提升。由于TS在试剂空间操作,因此无需枚举完整的库。随着库接近数十亿甚至数百亿个分子,TS相对于其他需要对完整库进行枚举和推理的AL方法具有优势。在本文中,首先介绍TS方法及其理论基础。接着,提供TS在三种不同虚拟筛选场景中的实际应用。
方法
TS是多臂老虎机问题25的一种算法,它有效地平衡了探索与利用。一个代理根据动作是最优的可能性依次采取行动。代理观察所选动作的结果(奖励),更新其对执行该动作的“多臂老虎机”的信念(belief),然后用更新后的信念(belief)重复这一过程。在TS中,维护了一个表示每个可能动作预期奖励当前信念的分布。在每一步i,代理从每个动作预期奖励的信念分布中随机抽取一个值,通过在这些随机样本中取最大值(或最小值,取决于优化目标)来选择一个动作,观察所采取动作的结果,并对该动作的信念分布进行贝叶斯更新。这个更新后的分布成为该动作新的信念分布,然后重复这一过程。
当使用TS搜索虚拟库时,我们对每个反应成分分别执行单独的TS计算。例如,在一个简单的两组分耦合反应中,我们会同时运行TS以选择第一反应成分的试剂,然后运行TS以选择第二反应成分的试剂。从第一个试剂的角度来看,第二个试剂的选择产生了打分值分布。一个动作包括为每个成分选择试剂,并用这些试剂制造一个分子(计算机模拟)。该分子通过期望的打分函数(如3D重叠或对接)进行打分,所观察到的分数被视为奖励。最后,根据观察到的分数更新用于制造该分子的每种试剂的预期奖励信念分布,并迭代地重复这一过程。
使用TS进行虚拟筛选有一些要求。首先,库必须包含反应和试剂,而不是未结构化的分子列表。第二个要求是,用于筛选分子的打分函数相对于被筛选库的大小和可用计算资源而言必须足够快。例如,对含有十亿个分子的化合物库使用打分函数进行TS需要一天的时间,则会因为成本或资源限制而变得不切实际。对库的1%进行采样将需要约27,000 CPU年(公式1)的计算能力。然而,使用相对较快的打分函数(即,大约需要1到10秒)可以在2800 CPU小时(公式2)内对十亿个分子的化合物库运行TS;相比之下,完全枚举则需要1 CPU年(公式3)。
若一个打分函数需要1天时间完成对含有1×109个分子的化合物库(假设采样库的1%)运行TS,近似所需时间为:
$$
{1\% \over 100} \times 1 \times 10^9\ calculations \times { {1\ day/calculation} \over {365\ days/year} } \times {1 \ CPU} \approx 27,000\ CPU\ years \cdots(1)
$$
若打分函数处理1个分子的时间为1秒,对含有1×109个分子的化合物库(假设采样库的1%)运行TS,近似所需时间为:
$$
{1\% \over 100} \times 1 \times 10^9\ calculations \times { {1\ s/calculation} \over {3600\ s/h} } \times {1 \ CPU} \approx 2800\ CPU\ h\cdots(2)
$$
若打分函数处理1个分子的时间为1秒,对含有10亿个化合物的完整库进行全枚举,近似所需时间为:
$$
1 \times 10^9\ calculations \times { {1\ s/calculation} \over {3.15\times 10^7\ s/year} } \times {1 \ CPU} \approx 32\ CPU\ years\cdots(3)
$$
当处理由反应组成的库时,TS一次只针对一个反应进行(虽然如果库中存在多个反应,这些反应可以并行搜索)。反应中允许的组分数量没有限制。然而,反应中某一给定组分(例如R1)中的任何试剂必须能够与该反应其他任意组分(Rn)中的任何试剂组合以形成有效的分子。
虽然TS算法在概念上可以适用于任何形式的信念分布,但某些形式会使信念分布的更新变得简单得多。我们选择假设与每个试剂相关的分数都遵循具有已知标准差的正态分布。
$$
𝑋_𝑖∼𝑁(𝜇_𝑖,𝜎^2)
$$
其中i是试剂的索引,\(X_i\)是该试剂分数的随机变量,\(μ_i\)是真实(但未知)的均值,σ是已知的标准差。我们将在下文讨论这一已知标准差的来源。根据这一假设,一个试剂的信念分布可以由时间t时\(μ_i\)的信念分布来表示,我们也假设这种信念分布是正态分布的。
$$
𝜇_𝑖∼𝑁(𝜇_{𝑖,𝑡},𝜎^2_{𝑖,𝑡})
$$
其中\(μ_{i,t}\)和\(σ^2_{i,t}\)是试剂i在时间t的索引。
在这些假设下,通过观察\(x_{i,t}\)来更新信念分布是非常简单的。
$$
𝜇_{𝑖,𝑡+1}={{𝜎_{𝑖,𝑡}^2𝑥_{𝑖,𝑡}+𝜎^2𝜇_{𝑖,𝑡}} \over {𝜎_{𝑖,𝑡}^2+𝜎^2}}\cdots(4)
$$
$$
𝜎_{𝑖,𝑡+1}={{𝜎_{𝑖,𝑡}^2𝜎^2} \over {𝜎_{𝑖,𝑡}^2+𝜎^2}}\cdots(5)
$$
实际上,我们发现正态分布对于表示诸如对接和3D叠合等标准打分函数非常有效。图1显示了来自Enamine REAL SPACE中的一个反应的单个试剂完全枚举的对接、ROCS和Tanimoto相似性打分值的分布,并叠加了正态分布的概率密度函数(PDF)。尽管这些分数的范围是有限的,不像正态分布那样,但是分布尾部的差异似乎并没有实际影响。
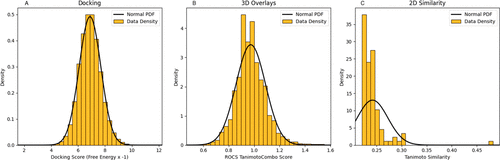
图1. 反应中随机选择试剂的枚举打分密度图,叠加了黑色的正态分布(A)对接打分、(B) 使用OpenEye的ROCS3D叠合的TanimotoCombo打分、以及(C) 2D相似性Tanimoto打分。
我们使用了一个简单的随机热身程序来产生初始的信念分布。这是通过为反应中的每个试剂制作n个随机分子并打分完成的,其中n由反应中组分的数量决定。对于双组分反应,我们发现n=3效果很好;对于具有\(>2\)种组分的反应,我们发现n=10是足够的。对于在热身过程中观察到的所有分数,我们计算经验平均值x̅W和经验标准偏差σ̅W。我们将已知的标准差以及所有试剂的信念分布设置为这些值。
$$
𝜎^2=\overline{𝜎}_w^2 \\ 𝜇_{𝑖,0}=\overline{𝑥}_w \\ 𝜎_{𝑖,0}^2=\overline{𝜎}_w^2
$$
然后,我们使用预热期间观察到的试剂打分值对每个试剂进行上述信念更新。
这种信念分布的先验设置本质上是从所有试剂都等价且等于热身期间观察到的完整分布开始。Xi的真实标准差可能低于在热身期间观察到的经验标准差,因为给定试剂的打分很可能比所有试剂中看到的更紧密,但我们仍然发现这个已知标准差的选择实际上可行。
接下来重复下述步骤:
- 对于每个试剂,从该试剂的平均值信念分布\(N(μ_{i,t},σ_{i,t}^2)\)中随机采样一个值。
- 每个反应组分(R1-Rn)试剂的选择,是根据每个组分采样值的最大或最小值(取决于打分目标)来为每个反应成组选择试剂。
- 用选定的试剂合成分子虚拟合成出分子,如果打分函数需要还进行构象分析
- 对分子打分
- 对信念分布执行贝叶斯更新(方程4和5)
跟踪过程中的打分靠前的n个分子(通常为100-1000)及其打分值。
重复上述步骤,直到达到停止标准:
- 库中的所有分子都被采样(即,完全枚举,通常仅适用于小型反应)
- 达到最大迭代次数(通常设置为库大小的0.1到1%,包括预热迭代)
请注意,确定最佳的停止标准仍然是一个需要研究的问题。
结果与讨论
用Tanimoto相似度搜索评估TS
作为该方法的初步评估,我们使用TS来识别大型组合库中最接近特定查询分子的化合物。在本次评估中,我们比较了TS与穷尽的Tanimoto相似性搜索的性能。相似性计算使用了RDKit(Version2023.03.1)2048位、半径为2的指纹。我们探究了在评估组合库时只考虑一小部分的情况下,在与查询分子最相似的100个分子中有多少个可以被识别出来。对于这个例子,我们考虑了一个基于Niementowski喹唑啉合成26反应组合的库,如图2所示。与此反应相关的试剂池包含了376种氨基苯甲酸、500种伯胺和500种羧酸。在这种情况下,完全枚举的库包含了9400万个产物分子。
图2. Niementowski喹唑啉合成路线
从喹唑啉库中随机选择了一个结构,如图3所示,作为查询分子。我们故意从组合库中选择查询分子,目的是为了有一个可重现的算例,模拟通过相似性搜索找到“令人感兴趣的”苗头化合物的场景。我们从Brown的2023年综述27中选出10个苗头化合物进行了同样的尝试,发现穷尽搜索找到的最相似分子的Tanimoto系数在0.23和0.34之间。这些Tanimoto系数并不落在我们通常认为需要跟进的范围。喹唑啉数据集中有两个分子与查询、查询本身及其对映体的Tanimoto系数为1.0。将查询与所有9400万个产物分子进行穷尽的Tanimoto相似度计算总共需要144个CPU小时。
图3.用于搜索三组份组合库的查询分子。查询分子的SMILES为CCc1cccc2c(═O)n(C3CNC3)c(C@@HN)nc12
为了高效地运行TS,我们需要了解应该从库中抽取多少分子来识别最佳分子。为调查这个问题,我们进行了2000到100,000次迭代之间的单独TS运行我们在2000到100,000不同的迭代次数之间进行了多次试验,并且收集了数据来分析结果。每个TS运行都执行了两次,一次是3个预热循环,另一次是10个预热循环。计算的结果如图4所示,图4-上的条形图显示了打分靠前100个分子的Tanimoto系数,作为比较,穷尽搜索打分的如右侧绿色所示。TS打分与穷尽搜索打分相比非常有利。即使在少量迭代的情况下,TS也能找到高分分子。此外,无论是3个还是10个预热循环的TS都能识别到与查询相似的分子。图4-下的柱状图显示了每次TS回收了多少个打分靠前的100个分子。当迭代次数不超过10,000时,我们可以看到使用10个预热循环的TS运行回收率略有增加。当我们移动到50,000和100,000次迭代时,我们没有看到预热循环数量的影响。而且,在50,000和100,000次迭代之间也没有发现任何差异。经过仅占总库0.1%的100,000次迭代后,我们可以回收到打分靠前100个分子中的90个。一个100,000次迭代的TS在一个CPU上花费了1.27分钟。
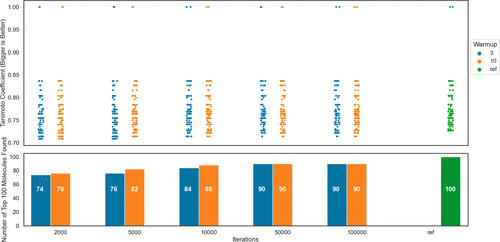
图4. 迭代次数对TS性能的影响。上图中的条形图显示了100个打分最高分子的Tanimoto系数。在最右边“ref”列,显示了从9400万个分子中的穷尽搜索得到的打分值。下图中的柱状图显示了随着迭代次数增加而回收到打分前100个分子的数量。
在TS和穷举搜索中,我们生成了打分值分布。确定前100个打分值的平均值是否存在统计学上的显著差异非常重要。为了比较使用不同预热计划的打分值分布,并将TS与穷举搜索进行比较,我们使用了学生氏t检验(Student’s t-test)。我们使用t检验来比较图4所示的所有分布对,并计算p值以确定我们是否可以拒绝均值等效的空假设(null hypothesis)。虽然0.05是一个广泛接受的显著性阈值,但我们必须考虑我们在执行多重比较。当执行多重比较时,拒绝空假设的概率会增加。因此,我们必须纠正显著性的阈值。调整多重比较的一种方法是Bonferroni校正,它通过显著性阈值除以N,其中N为比较次数。对于图4的情况,其中比较了11个TS计算结果,因此阈值为0.05/11或0.0045。
图S1显示了来自t检验的所有分布对的p值热图,这些分布对在图4上半部分中所示。任何比较的最小p值为0.056,这是指穷尽搜索的参比(ref)与2000次迭代、三个预热循环TS之间的比较。其余的p值范围在0.15到1.0之间。我们的结论是,在每种情况下,前100个分子的平均打分值没有显著差异。
为了评估TS程序的可重复性,进行了两组不同的10次独立的TS计算(总共20次)。两组之间的唯一区别在于热身阶段是如何进行的。在第一组中,每个试剂都与三个随机配对试剂一起采样。在第二组中,采样的配对试剂的数量增加到10个。在上述研究的指导,每项TS使用了50,000次迭代,搜索了总库的0.05%数据。在图5-上中,再次看到了打分靠前100个分子的Tanimoto系数条形图。“concat”列显示了将10次TS合并起来并选择打分最高的100个不重复分子的结果。在所有的TS中,观察到了类似于穷尽搜索中的打分靠前100分子的打分值分布,如图表右侧绿色条形图所示。在图5下,TS始终回收了打分靠前100个分子中的88~90个。如上所述,我们在图5上对所有打分值分布对进行了统计分析,结果见图S2中。在这种情况下,我们无法建立任何一对分布之间具有统计学意义的差异。在所有的比较中,最差的p值为0.64。
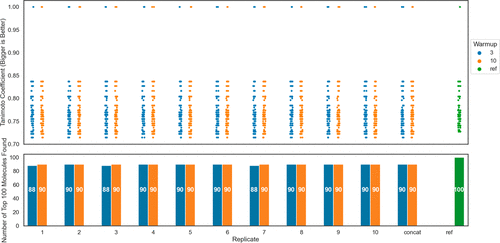
图5. TS过程可重复性的评估。上图显示了100个打分最高分子的Tanimoto系数,右侧绿色的结果是穷尽搜索评估的结果。图下的条形图显示了10次重复TS运回收打分靠前100个分子的数量。如图4所示,比较了3和10个预热循环。
为了给TS提供基准比较,我们与随机选择进行了对比,从图2中突出显示的喹唑啉库中抽取了50,000个分子。如上所述,我们评估了TS搜索回收到的打分靠前100个分子的数量,并将其与使用50,000个随机样本回收到的分子数量进行比较。对于TS,我们使用了10个预热循环并在50,000次评估后停止搜索。TS和随机选择分别重复10次,以图3中的查询分子的Tanimoto相似性作为目标。图6展示了比较结果,采用了上述图表的形式。在图6上中,红色与橙色分别表示的是随机选择与TS选择的打分靠前100个分子的Tanimoto系数。右侧的绿色点代表穷尽搜索得到的打分靠前100个分子的打分值。图6下的柱状图显示了使用TS和随机搜索回收到的打分靠前100个分子的数量。正如上面描述的,“concat”列代表所有10个TS的合并产生的前100个分子。在10种情况中有9种情况下,随机搜索没有回收到任何打分靠前的100个分子。在第7次重复中,随机搜索回收到了打分靠前的100个分子中的一个。
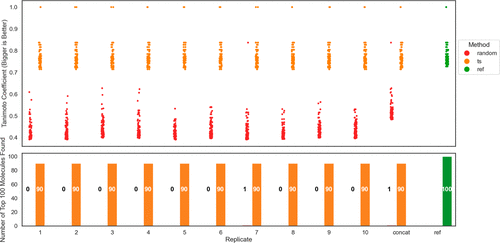
图6. TS与随机搜索的比较。上: 显示了100个打分最高分子的Tanimoto系数,右侧红色显示的是随机搜索的结果,橙色是TS的结果,绿色是穷尽评估的结果。下:条形图显示了在10次重复TS中回收到前100个分子的数量。在10次重复中有9次,随机搜索未能回收到任何前100个分子,因此,红色条形不可见。
为了拒绝图6中分布的平均值等价于空假设,我们进行了类似于上述描述的统计分析。该分析的结果如S3图所示。在这种情况下,我们看到TS和随机选择之间存在显著差异,p值小于10-100。11个TS重复之间的差异并不显著。
TS错失了什么?
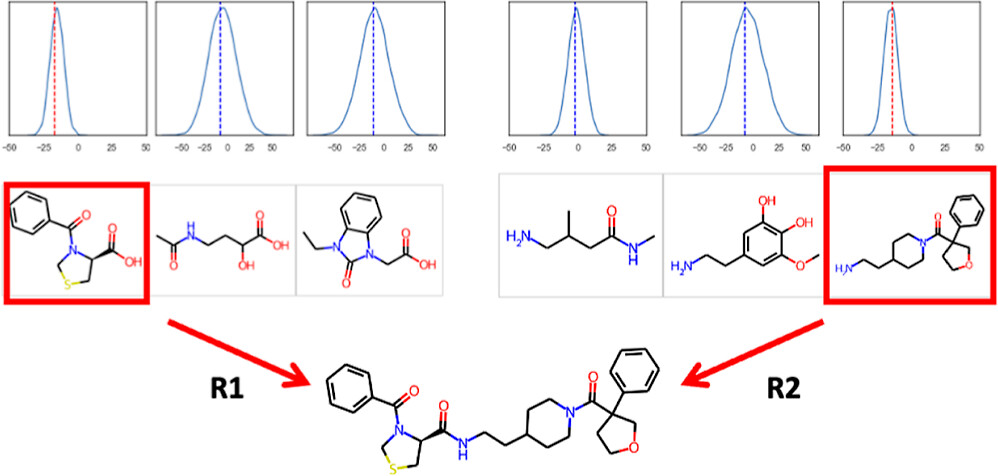
在上面给出的例子中,TS从Tanimoto相似性搜索中识别了打分靠前100个苗头化合物中的90个。此外,合并多个搜索的结果影响很小。在大多数情况下,TS在所有10次重复中都识别到相同的苗头化合物。为了更好地理解TS的性能,我们编制了表2,该表格显示了打分靠前100个苗头化合物所包含的砌块。根据穷尽的搜索结果,前100个分子由5种R1、12种R2和24种R3砌块组成。除了展示结构外,表2还指出了砌块在前100中的出现频次。在每个砌块结构下,我们展示了含有特定砌块的分子数量以及TS发现的实例数。例如,在左边的R1砌块被包含在前100个分子中的92个中,并且92个实例中有90个被TS找到。因此,我们在结构下方列出“90/92”。
表2. 用于生成Tanimoto相似性示例中打分最高分子的砌块a
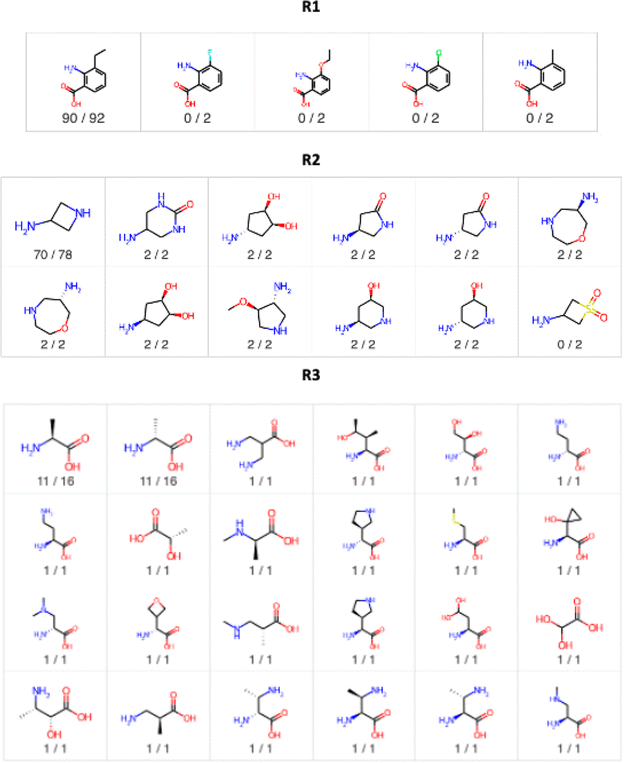
a.在每个结构下面的分数中,分子表示TS发现该试剂出现的次数,而分母则表示该试剂在前100分子中出现的频次。
查看表格2,我们看到虽然TS只识别出了五个R1砌块中的一个,但这个砌块占前100个分子的92%。这一结果指出了我们当前TS实现的一个潜在局限性。当一个砌块对分数贡献显著时,在该位置的解决方案的多样性可能会受到限制。可以采用多种方法来增加采样的多样性,包括限制砌块被采样次数或引入类似于Smellie28用于构象采样的投票方法。提高采样效率是我们团队正在积极研究的领域,我们希望在之后的文章中进一步报告进展。通过发布我们的开源TS代码,我们也希望其他人能够开发改进的采样方法,并公布他们的方法和代码。尽管TS未能识别一些R1砌块,但对于R2和R3的结果更为乐观。对于R2,TS识别了12个打分最高砌块中的11个,仅未能识别第二行最右边的环丁基硫酮。对于R3,TS识别了所有的羧酸砌块。有趣的是,打分最高的两个R3试剂是d-丙氨酸和l-丙氨酸的外消旋体。由于这里运行的Tanimoto相似度搜索没有考虑立体化学,具有不同立体化学的甲基手性中心的分子将获得相同的Tanimoto评分。
TS与机器学习模型辅助的主动学习(AL)比较
在过去的几年里,机器学习模型辅助的主动学习(ML model-assisted AL)已经成为大规模虚拟筛选的一种流行方法。 在机器学习模型辅助的主动学习中,一个机器学习模型作为更昂贵方法计算(如对接或自由能计算)的代理。在迭代过程中,主动学习算法试图平衡化学空间的探索和特定有前途区域的开发。主动学习过程通常首先从较大的分子数据库中采样一个小子集,通常是几百到几千个分子。我们将这个子集称为训练集。 选择后,在训练集分子上进行昂贵方法计算,并对机器学习模型进行训练以预测来自训练集结构的昂贵方法计算值。然后使用该模型来预测DB中所有分子的昂贵方法计算的值。 这些预测值指导了下一组分子(S1)的选择用于昂贵方法计算。可以采用多种策略来选择S1。在“贪婪”的策略下,N个得分最高的分子被选为下一个轮次的昂贵方法计算。其他选择策略可以利用诸如预测中的不确定性等特征来平衡探索和开发。在S1上完成昂贵方法计算后,化学结构和相应的昂贵方法计算值添加到训练集中,并且用来训练一个新的模型、并用来预测DB的一组新的昂贵方法计算值。选择子集、昂贵方法计算、建立模型、在DB上预测以及选择另一个子集的过程会持续预定次数的迭代。
TS和AL之间有两个重要的区别:
1.AL需要对库进行完整的枚举,而TS不需要。对于大型库来说,这种枚举可能在计算上非常昂贵。上面描述的9400万个分子库几乎花了单个CPU 7个小时来穷举。当一个数据库接近十亿个分子时,所需的枚举时间可能会变得冗长。虽然已枚举的库可以存储在磁盘上并重复使用,但数十亿个分子的库将消耗几百GB的磁盘空间。
2. 模型预测必须在每个AL循环中对DB中的每个分子进行计算。当库变得非常大时,这可能需要很长时间。假设机器学习模型可以每秒对1万个分子执行推理,则对于十亿个分子的预测将需要一个AL循环27小时。此外,十亿个分子的描述符通常不适合计算机内存,因此必须采用并行化或分页策略。
虽然通过并行处理可以提高AL中枚举和推理的速度,但这些改进是以计算架构显著复杂化为代价的。
为了比较TS和AL的计算可行性和可重复性,使用了图2中喹唑啉库的缩小版本。在这个例子中,R1、R2和R3试剂各使用了100个,形成了一个总共包含一百万个分子的库。为了使TS和AL的评估数量相等,每种方法总计进行了5000次评估。出于比较的目的,在TS部分,在5000次评估后停止。对于AL,进行了五个循环,每个循环有1000个分子,总共进行了5000次评估。与上面的重现性分析一样,TS和AL都分别重复了十次。
TS按照上述方法进行,包括10个预热循环。对于AL,我们最初采样了1000个分子,并计算这些分子与图3分子之间的Tanimoto相似度。然后用Tanimoto相似度和这1000个分子的结构来构建一个ML模型。在该模型中,分子表示为半径为2的2048位Morgan指纹。随机森林回归(通过scikit-learn版本1.2.2实现)用于建立预测Tanimoto相似性的模型。然后将此模型应用于总共一百万分子数据集的推理。在贪婪选择过程中,将在前一轮未选中的1000个打分最高的分子添加到训练集中,并重新训练模型。这个过程总共重复五次。与此论文相关的GitHub存储库提供了一个带有AL实施的Jupyter笔记本。
图7显示了TS和AL之间比较的结果,采用类似于图4-6的格式。在顶部的条形图中,我们看到TS和AL的打分分布相似。使用学生氏t检验来穷尽比较图7顶部图表中所有分布的均值,唯一显著差异是AL第6次重复和其他重复之间的差异。具有完整p值集的热图可用于成对比分析,结果见图S4。图7底部的柱状图显示了每次重复中由TS和AL找到的打分最高100个苗头化合物的数量。TS找到了打分最高100个苗头化合物中的79~84个,而AL则找到了61~100个。这表明基于整个分子表示构建模型的AL方法的优势。然而,鉴于该库规模较小并为单一实例,这很难得出一般性的结论。如上所述,目前在试剂空间中进行搜索的方法有一些局限性。需要进一步的研究以确定TS的最佳协议,并确定TS与其他方法最合适的比较方式。
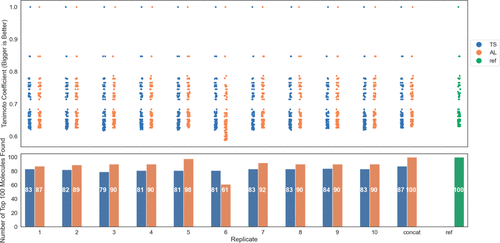
图7. TS和模型辅助主动学习(AL)的比较。顶部图形中的条形图显示了打分最高100个分子的Tanimoto系数。“concat”列显示了左侧10次TS/AL合并后打分最高100个分子的打分。最右侧的“ref”列显示了一百万个分子的穷尽搜索结果。图下部的柱状图显示了每个重复回收到的打分最高100个分子的数量。
到目前为止,我们一直在用简单的相似性搜索来展示TS方法的实用性。虽然这提供了一种快速评估TS的方法,但当更昂贵的计算方法,如对接或3D相似性搜索用作目标时,该技术的实际价值变得明显起来。我们的开源TS实现定义了一个称为“评估器”的抽象基类,它为TS指定评估函数。从评估器基类继承的Python类可以使用各种标准,包括3D相似性、对接打分、3D相似性和机器学习模型。评估器类接受RDKit分子作为输入,并返回一个浮点数打分。在Python类中定义TS过程时,用户可以定义更高/低的打分值是否代表更好的结果。
用TS对2.34亿个分子进行3D相似性搜索
为了评估TS在3D相似性搜索中的能力,我们使用OpenEye的ROCS29进行了搜索,计算使用OEShape Toolkit (Version 3.4.11)进行。ROCS叠合分子构象,并根据原子中心的高斯表示法的重叠来计算打分值。ROCS输出两个在0和1之间的分值。其中形状打分评估两个分子的空间重叠,颜色打分评估相应的药效团重叠。这两个分值之和,在0到2之间,称为TanimotoCombo打分。在这个例子中,我们使用了一个基于反应m_22bbh的库,如图8所示,来自Enamine REAL集合。该库包含13,995个仲胺和16,724个羧酸,总共产生了234,052,380个反应产物。

图8. 用于ROCS和对接实验的Enamine反应M22。
与Tanimoto相似性算例一样,从库中选择一个随机分子作为查询式(query),如图9所示。在库中有两个分子与Query的TanimotoCombo打分为2.0,分别为该分子本身及其对映异构体。
图9. 用于ROCS搜索的查询分子
对这个库进行穷尽的ROCS搜索需要110,208个CPU小时(12.5个CPU年)。我们使用ROCS打分作为TS的目标函数,搜索同一组合库的0.1%。在本算例中,我们用了10个预热循环。每次TS运行需要32个CPU小时。进行了10次TS之后,将这10次TS合并,取不重复的打分最靠前100个分子,在图10中标记为“Contat”组。图10遵循与上面图4-7相同的格式。在本算例中,图上显示了来自10次TS运行、合并数据和穷尽搜索的打分靠前100个分子的TanimotoCombo打分。图10的上图表明每个TS都识别出许多得分最高的分子。图10下进一步用每轮中识别出的打分前100个分子的数量来量化了说明这一点。在表现最差的TS中发现了其中54个最佳分子。10次TS的合并识别出了100个最佳分子中的69个。虽然ROCS结果没有显示出我们在Tanimoto相似性搜索中看到的同样的加和性能,但我们只需评估库的0.1%就可以识别出最佳100个分子的54~69%。
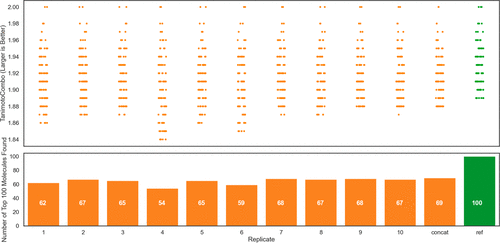
图10. 对2.34亿个分子进行TS与穷尽ROCS搜索的性能对比。上:显示了前100个分子的ROCS打分分布,“concat”列显示了左侧10次TS合并后打分最佳的100个不重复分子的打分,“ref”列显示了穷尽搜索的结果。下:条形图显示了TS回收到在穷尽搜索中打分靠前100个分子的数量。
图S5显示了10次TS ROCS重制和穷尽搜索参比的打分分布的成对比较。穷尽参比与TS第6次重复之间的成对比较p值最低(p=0.01)。鉴于我们正在比较11个ROCS,因此最低p值大于Bonferroni校正阈值0.0045。
用TS对接3.35亿分子
作为TS如何实际应用的另一个例子,我们检查了一个内部蛋白质靶标的对接研究。在本算例中,我们用Enamine库m_22bba比较了TS与穷尽对接的性能。该库用图8中定义的反应但不同的砌块集合构建组合库。试剂由17,741个仲胺和18,873个羧酸组成,生成了334,825,893个产物。对接使用OpenEye的OEDocking工具包版本(4.0.01)30,31。对接打分函数为ChemGauss4,其结合了经验性和基于物理的打分项。蛋白结构用OpenEye Spruce工具包版本1.4.0.0中的默认设置准备。对接的活性位点用一个尺寸为20×23×19 Å、中心位于结合位点上的盒子来定义。在亚马逊网络服务上使用10,000台云CPU运行穷尽搜索,消耗了160万CPU小时(1826 CPU年)。为了评估TS的表现,用我们的实现来搜索相同的3.35亿分子。根据我们的研究,使用TS进行对接需要比上述ROCS或Tanimoto搜索更多的采样。因此,我们设定迭代次数设定使得库的1%被搜索到。
我们将10次TS的结果与通过整个库的穷尽对接获得的真实结果进行了比较,并评估了TS识别穷尽搜索中打分最高100个分子的能力。在图11-上图中,我们绘制了每个10次TS打分最高100个分子的对接打分。为了与前面的图表保持一致,将ChemGauss4分数乘以-1.0使得值越大表示分数越好。如上所述,“concat”列显示了将左侧10次TS结果合并取不重复的打分最佳100个分子的对接打分。。在图10-下图中,我们可以看到所有10次TS重复都能识别出一半以上的前100个分子。在最糟糕的情况下,TS可以识别出前100个分子中的57个。对10次结果合并后,回收到的前100个分子的数量增加到了62个。图S5展示了图11-上图分布的成对比较的p值。虽然10次重复之间的差异并不显著,但TS和穷尽参比之间存在显着差异(最小p=0.00033)。
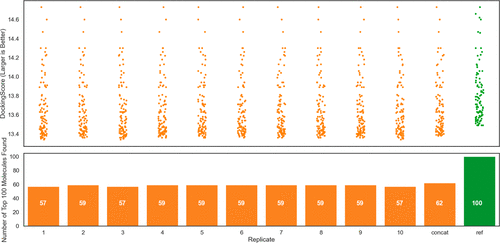
图11. 对3.34亿个分子的TS与穷尽对接性能比较。上: 显示了打分靠前100个分子的对接打分分布,“concat”列显示了左侧10次TS合并后打分靠前100个不重复分子的打分值,“ref”列显示了从穷尽搜索得到的结果。下:条形图显示了通过TS回收到的穷尽搜索打分靠前100个分子的数量。
TS的工作原理与内部细节
图12显示了对接、3D叠合和Tanimoto相似度穷尽搜索打分的背景分布。每个图中的蓝色垂直虚线表示TS识别出的打分靠前100个分子的最低分值,及其Z-score。高Z-score(范围为6.7-12.5)表明TS高效地对分布的极端尾部进行了抽样。
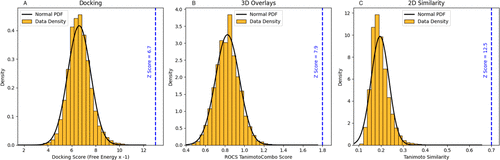
图12. 在使用(A)对接,(B)ROCS和(C)Tanimoto相似性进行穷尽搜索时每个库的背景分布。这些分布是通过从穷尽搜索中完整的打分数集中随机采样10,000到50,000个打分创建的。背景密度显示为橙色,并叠加了一个正态分布PDF。垂直蓝色虚线代表当对同一库TS时,在打分靠前100个分子中确定的最低打分;垂直线的Z-score以蓝色文本表示。
在我们评估参数设置对TS性能影响的时候,检查与单个试剂相关的信念分布的演变是有益的。在图13中,我们绘制了Tanimoto相似度、ROCS和对接方法打分靠前100个产物相关试剂的信念分布。在三个算例中,我们都使用相同的1百万分子库进行TS评估,该库是通过quinazoline反应构建的,如图2所示,从100个R1、100个R2和100个R3试剂构建的。用于ROCS和Tanimoto相似度计算的查询分子结构如图3所示。在本算例中,查询分子不包含在被搜索的1百万分子中。对于对接评估,将这些分子对接到JNK3(PDB 2ZDT)的正构位点( orthosteric site)。TS的参数类似于上面列出的那些。所有TS总共进行了10次预热循环。在每种方法打分靠前100个分子中的不重复试剂的数量显示在表3中。正如预期的那样,“更直接”的Tanimoto相似度计算主要由少数R1和R2试剂主导。在“更抽象”的ROCS和对接计算中,更多的试剂出现在打分靠前的100个分子里。
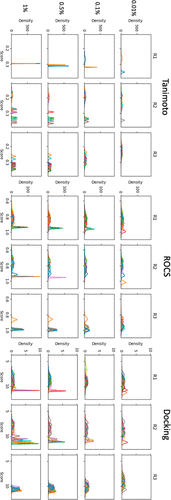
图13. 包含百万分子库中前100个分子的试剂的信念分布。图中的行对应于已搜索的库的百分比。
表3. 对百万分子库的Tanimoto相似性、ROCS搜索和对接计算后,在打分靠前100个产物分子中不重复试剂的数量
| Method | R1 | R2 | R3 |
|---|---|---|---|
| Taimoto | 2 | 5 | 23 |
| ROCS | 34 | 6 | 12 |
| Docking | 24 | 9 | 41 |
图13中的每个子图显示了构成打分靠前100个产物分子的试剂的核密度估计(KDE)。该图包括三个子图,分别展示了R1、R2和R3的KDE。图中的行表示采样的数量逐渐增加。在第一行中,已经对100个分子(占总数的0.01%)进行了采样。在接下来的几行中,分别对1000、5000和10,000种分子进行了采样,这些分子分别占总数量的0.1%、0.5%和1%。在检查图13的第二行时,我们看到,在对数据库采样0.1%之后,对于Tanimoto相似性和ROCS出现了领先的试剂。但是,我们也看到了对接计算的信念分布都非常相似。虽然R2有一些右移的对接分布,但R1和R3之间的差异非常小。当我们接近搜索数据库的0.5%并最终达到1%时,我们会看到R1和R2领先试剂的出现。对于所有三种方法,我们发现R3分布的分化较少。对于Tanimoto相似性和ROCS,查询分子中的R3基团仅包含三个原子。对于对接,探索的R3基团涵盖了多种疏水性和氢键相互作用,并且许多R3基团打分相似。除了提供有关TS内部运作的见解外,像图13这样的图表使用户能够快速确定打分靠前分子的哪些部分具有一致的高分相互作用。
建议与未来研究方向
我们的研究证明了TS在虚拟筛选任务中广泛用途。基于经验研究,我们发现两个组份库通常需要三个预热循环,而三个组件库则更偏好十个预热循环。然而,由于预热循环占总体运行时间的一小部分,我们将通常将此参数设置为10。我们倾向于不设定预定义的迭代次数。相反,在未到预定义的迭代次数(根据任务的不同,通常是1000到10,000)而打分却没能继续改进时,我们会停止TS计算。最终,将TS应用于虚拟筛选是一种新技术,最佳实践必须通过实验来发现。鉴于TS的速度,可以快速执行参数扫描,并且该方法可以根据特定目标和数据集进行调整。我们希望通过发布我们的TS实现代码,其他人会抓住机会探索参数并报告他们的经验。
当然,TS也有一定的局限性。首先,它只能应用于已知的库化学和砌块的组合库。目前无法将TS应用到未指定库化学的多样筛选库中。可以想象使用逆合成来分解一个库,并在砌块上使用TS,但这并不容易实现。另一个限制在前面的已经指出。在某些情况下,TS可能会过于关注特定的砌块而错过打分最高的产物。如前所述,有许多方法可以改进采样并整合额外的化学智能。即使有这些限制,TS仍是一种有用的技术。本文的例子表明,通过搜索不到百分之一的数据集,TS可以识别出一半以上的高分分子。
我们的TS应用与标准的多臂老虎机框架在几个方面有所不同。首先,打分分布是非静态的。从单个试剂的角度来看,其他试剂的信念分布更新将产生随时间变化的分数分布。此外,一旦选择了给定的一组试剂,我们永远不会再次评估该组试剂。一旦为给定试剂挑出最佳伴侣,此试剂的未来打分将会下降。其次,我们的目标是找到联合分布的尾部(最高分分子),而不是找到与其他试剂配对时具有最佳平均打分的试剂(这是典型的TS后悔目标)。最后,我们通常想要找到一组高分分子,而不仅仅是最好的试剂组合。典型评价的TS会建议立即找出单一的最佳试剂,并仅选择这些试剂。
尽管与TS理论基础的存在差异,但这里显示的经验性能是强大的。然而,我们确实观察到算法有时无法找到所有打分最高的分子,如图4、5、10和11中的平台所示。我们认为这可能是由于在评估TS的理论框架和基于试剂的虚拟筛选的实际应用之间存在差异所致。对理论分析或算法进行扩展以更好地解决这些差异是未来工作的方向。
在本研究中,我们展示了TS的应用,其中分子被逐一枚举并打分。然后使用整个分子的打分来更新相应砌块的信念分布。TS也可以以批处理模式运行,在这种模式下,一组分子会被枚举和评估。为批处理模式下的TS开发最佳规划是一个有待研究项目,我们希望在未来对此进行更多报道。
结论
TS提供了一种高效的搜索超大型组合库的方法,该方法因按需化学合成的增加而变得普遍。这种高度灵活的方法可以应用于各种目标,包括2D-和3D-相似性搜索以及蛋白质-配体对接。原则上,使用任何将分子作为输入并返回打分值的功能都可以应用TS。该方法运行的唯一要求是必须有一个可以组装成最终分子的砌块库,并且最终可以进行打分。
有了TS,我们可以评估数据集的不到1%,找到最佳分子的重要部分。表4比较了本文中三个算例TS和穷尽搜索所需的CPU时间。在我们的测试中,TS实现了比穷尽搜索快了1000多倍的速度提升。使用多个CPU,我们可以在几个小时内搜索十亿个以上分子的化合物库。根据评价指标的不同,TS可以识别出从穷尽搜索打分靠前100个分子中的57~90个。这种速度和准确性的结合使得TS成为虚拟筛选工具箱的一个绝佳补充。
表4. 此处给出在三个算例中TS和穷尽搜索运行时间的比较
| Items | molecules searched | exhaustive CPU hrs | TS CPU hrs |
|---|---|---|---|
| Taimoto similarity | 94,000,000 | 144 | 0.003 |
| ROCS search | 234,000,000 | 110,208 | 32 |
| Docking | 335,000,000 | 1,600,000 | 16,000 |
代码
为了便于重现、扩展本研究的内容,在https://github.com/PatWalters/TS提供了TS的开源参考实现。用于Tanimoto验证的喹唑啉库可在同一存储库中获得。由于许可限制,我们无法分发使用进行穷尽搜索的Enamine REAL库,但是REAL库可以从Enamine获取。虽然我们很想提供在开放库进行超大型库的TS和穷尽对接的比较,但我们不能证明穷尽搜索的费用是合理的。上述GitHub存储库包含Python类,可用于执行ROCS与对接的TS评估。这些组件需要OpenEye的软件许可证。将现有框架扩展到其他对接和分子相似性方法应该是比较简单的。
支持信息略
略
文献
略
在线视频
Pat Walters. Applying Active Learning in Drug Discovery
推荐阅读
- Hongtao Zhao, Eva Nittinger, Christian Tyrchan. Enhanced Thompson Sampling by Roulette Wheel Selection for Screening Ultra-Large Combinatorial Libraries. 2024https://doi.org/10.1101/2024.05.16.594622
在本研究中,阿斯利康Zhao等人在Relay团队的TS基础上,旨在解决其一些不足之处并提出优化方案。Zhao等人引入了一个预热程序,以确保以评估最少数量的分子为所有试剂设定初始概率。此外,还提出了一种带有自适应性停止标准的轮盘赌选择法,以提高采样效率,并且仅当试剂出现在新分子中时才更新其信念分布。作者证明了通过采样枚举库的0.1%,可以实现100%的回收率,展示了所提出优化方案的有效性。相关代码见:https://github.com/WIMNZhao/TS_Enhanced
联系我们
想在自己的项目中使用OMEGA、ROCS与OEDocking等OpenEye软件进行超大规模虚拟筛选,请联系我们获取免费的试用版;你还可以委托研究的方式使用该软件。
- 电邮:info@molcalx.com
- 电话:020-38261356
原创文章,作者:小墨,如若转载,请注明出处:《Thompson sampling──高效的超大型按需合成库虚拟筛选方法》http://blog.molcalx.com.cn/2025/02/12/thompson-sampling.html


