
摘要:生物等排体替换(Bioisostere replacement)是在药物发现中用于优化候选分子活性和选择性的一个强大且流行工具。选择正确的生物等排体以投入资源进行合成和后续优化,是高效药物发现项目的关键。在此,我们展示了如何将三维定量构效关系(3D-QSAR)与相对结合自由能计算结合到一个主动学习工作流中,以对包含数百个生物等排体分子库进行优先级排序。通过一个人类醛糖还原酶(aldose reductase)的测试案例,我们证明了使用该工作流可以快速定位结合最强的生物等排体替换,并且具有相对适中的计算成本。通过3D-QSAR与FEP的主动学习结合,在保证精度的同时将计算成本降低80%,为药物发现提供高效筛选工具。
原文:Ramaswamy, V.K., Habgood, M. and Mackey, M.D. (2025) “Active Learning FEP Using 3D-QSAR for Prioritizing Bioisosteres in Medicinal Chemistry,” ACS Medicinal Chemistry Letters. Available at: https://doi.org/10.1021/acsmedchemlett.4c00554.
编译:肖高铿/2025-01-05
前言
为了将苗头化合物转化为强效的先导化合物,从苗头化合物到先导化合物(hit-to-lead)的优化需要在活性、安全性、理化性质和知识产权之间进行微妙的平衡。生物等排体替换(bioisostere replacement)是一种广为接受并经常应用的方法,用于从起始分子(starting molecule)生成先导样化合物1。Spark生物等排体和R-基团替换软件2利用Cresset的静电和形状相似性方法3可用来为苗头化合物的一部分生成数百个可行的替换选项,但选择少数几个重点投入资源以提高药物化学/药物发现项目成功率的责任在于用户。虽然Spark打分算法在建议能够保持一定程度生物活性的替换方面表现良好,但这类对接和配体相似性方法通常无法准确预测确切的活性水平4-6。
有多种方法可用于更精确地根据蛋白质-配体结合亲和力对分子进行排序。金标准是严格的基于物理的方法,如自由能微扰(FEP)。然而,这些方法可能对算力要求非常高,特别是当考虑包含几百个分子的数据集时。诸如定量构效关系(QSAR)建模之类更为启发式的方法,允许更快的预测,但需要大量的训练数据,因此通常不适用于早期的苗头到先导阶段。在此,我们展示了这两种方法可以被链接成一个循环工作流,以结合两者的优势来提高其预测能力,并用它们来筛选最终的一组先导分子。这种工作流常被称为主动学习(active learning),它涉及使用机器学习模型来指导寻找排名靠前的分子,同时显著减少执行的FEP计算数量,以克服时间和成本相关的挑战7,8。
已有几篇文献评估和应用了不同的设计策略、数据集大小、活性来源以及主动学习迭代次数来将主动学习应用于自由能微扰(FEP)8–11。这些研究无一例外地使用了传统的基于分子指纹描述符的2D-QSAR。鉴于Spark算法专注于在三维空间中识别与目标分子具有高度静电和形状相似性的生物等排体替换,所有分子在一个给定的蛋白质环境中都是叠合良好的,并且具有高度的3D相似性。因此,所得到的数据集非常适合于3D-QSAR。在本文中,我们展示了利用3D描述符所提供的额外信息使得QSAR方法即使在少量训练数据的情况下也能更具预测性,从而使得在相对较少的活性评估次数下进行主动学习成为可能(图1)。
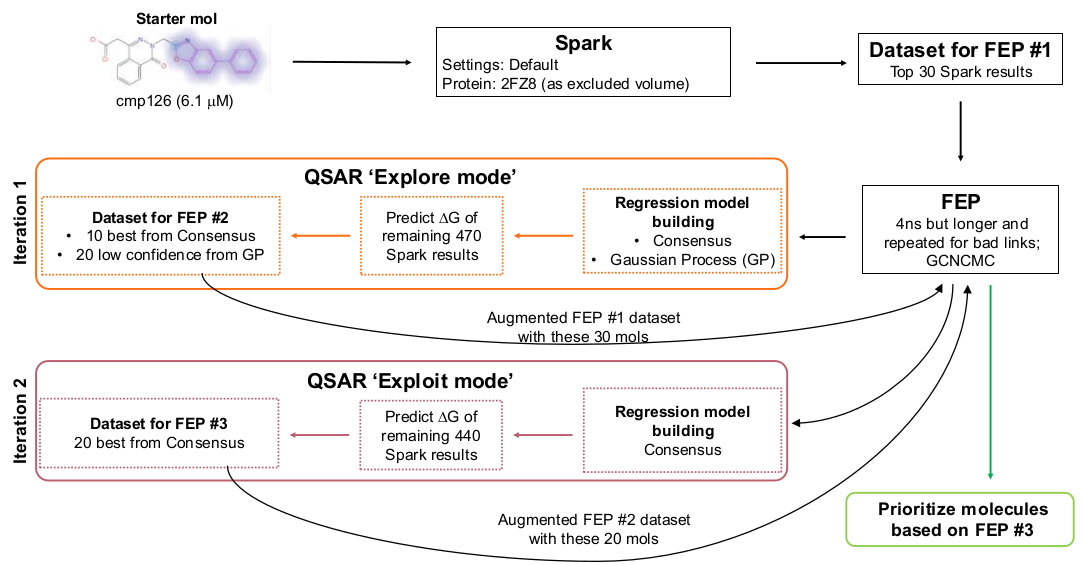
图1. 从苗头到先导的不同阶段应用3D-QSAR主动学习FEP的工作流示意图
糖尿病是一种慢性代谢性疾病,对全球有着显著的影响,以至于到2025年阻止糖尿病和肥胖症上升成为了全球公认的目际(https://www.who.int/health-topics/diabetes)。醛糖还原酶(aldose reductase,ALR2)在NADPH存在的情况下将葡萄糖还原为山梨醇,这是多元醇通路(polyol pathway)中的第一步也是限速步骤。在高血糖状态下,ALR2的过度活跃会导致细胞内山梨醇的积累、胞质NADPH的消耗以及氧化应激,最终导致并促进糖尿病并发症的发生和发展,如肾病、神经病变、视网膜病变、动脉粥样硬化及其他心血管疾病12,13。ALR2抑制剂被认为是治疗这些糖尿病并发症的一种有希望的工具,而众多的ALR2抑制剂构效关系数据14–17使其成为评估我们工作流的理想对象。
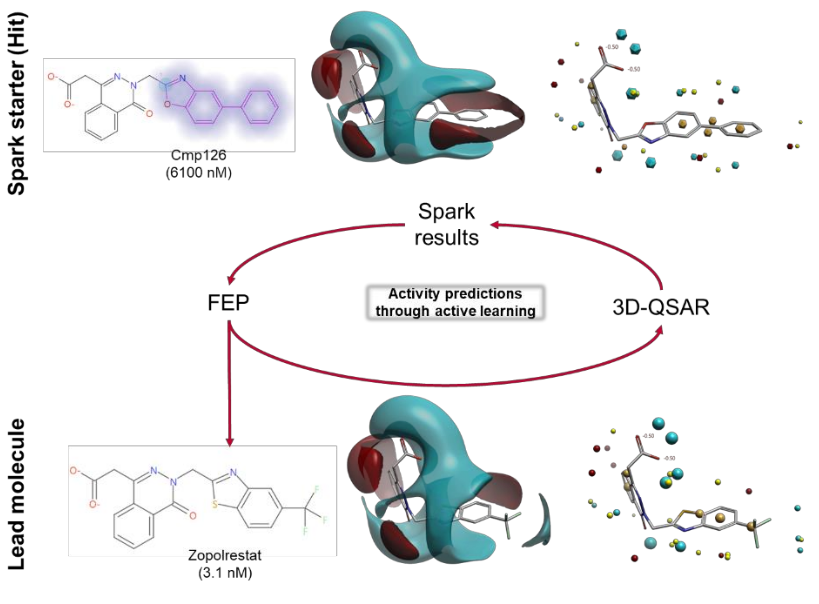
想象在一个典型的药物发现项目中的从苗头到先导阶段,我们选择了辉瑞公司报道的取代噻唑系列15中的一个低活性结合分子cmp126(pIC50=5.21)作为我们工作流的起始分子。我们使用cmp126作为R-基团替换实验的起始分子(见方法部分),通过Spark生成了苯并噁唑类(图2)的一系列生物等排体。
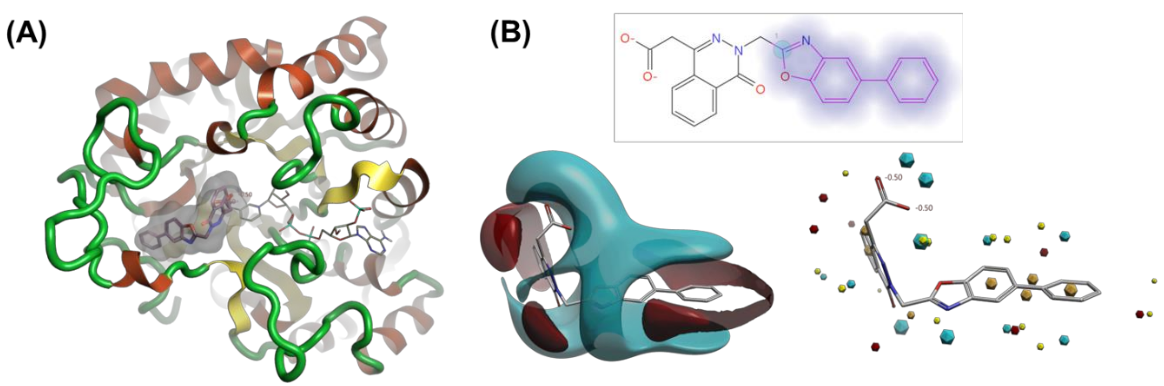
图2.(A)Zopolrestat(仅呈现为分子表面)与人类醛糖还原酶(呈现为飘带)的共晶X-衍射结构(PDB: 2FZ8),以及在Spark中的起始分子(见在B面板)嵌入其中(呈现为棍棒模型),同时展示了相邻的结合辅因子烟酰胺腺嘌呤二核苷酸磷酸。(B) 2D结构是起始分子(cmp126),对人类ALR2的pIC50值为5.21(即IC50=6.1μM)。高亮显示部分是在Spark中选定进行R-基团替换的区域。左下角和右下角分别展示了分子的三维分子静电势(MEP)和场点。MEP和场点的颜色代码:红色代表正静电场,蓝色代表负静电场,黄色代表形状,橙色代表疏水性。
Spark通过评估用生物等排体替换后的分子与起始苗头化合物之间的形状和静电相似性来对生物等排体进行打分。这种相似性评估是在“产物空间”中进行的,而不是在“片段空间”中,因为一个片段的静电性质可以因其所在的分子环境不同而显著不同。在这个案例中,Spark识别出了多个已知文献报道的苯并噁唑的替代物,包括苯并噁唑、苯并噻唑和苯并二氮杂卓取代的分子15,16。在建议的500个生物等排体替换中,共有32个ALR2抑制剂具有已报告的活性数据(pIC50范围从5.21到9)。虽然Spark在识别保持一定程度生物活性的生物等排体方面非常成功,但基于相似性的打分方法并不足以准确地对所建议的生物等排体替换进行排序(图SM2)。相对结合自由能方法可以提供显著更准确的排序,但是将这些方法应用于所有500个生物等排体建议将会产生相当大的计算成本。
主动学习协议的初始阶段需要为一组初始分子生成预测活性数据,以训练机器学习模型。文献中的协议通常建议这个初始数据集包含大约100个分子,但使用信息更丰富的三维描述符应该可以减少这一数量。因此,初始数据集选择了Spark结果中按相似性打分排序靠前的30个分子。按照标准的Flare FEP协议18,19(见方法部分),并标记cmp126作为具有已知实验活性的参比分子进行了研究。
然后利用FEP预测的这30个分子的结合自由能(ΔG)构建了两个3D-QSAR回归模型(见方法部分):共识(Consensus)模型和高斯过程(Gaussian Process)模型。这两个模型在交叉验证训练集上的评估指标均显示出良好的预测能力。共识模型和高斯过程模型的Q²值分别为约0.89和0.82,而它们各自的Kendall’s tau值分别约为0.79和0.74(见图SM 3A)。这两个模型随后被应用到数据集中剩余的470个分子上。对于主动学习的早期轮次,建议通过使用机器学习预测来最大化数据集的信息含量(所谓的“探索”模式)以获得最佳结果,而不仅仅是选择预测活性最高的分子。因此,我们根据共识回归模型挑选了排名前10的配体,并从高斯过程回归模型中选取了表现出最大标准偏差的20个分子。目的是为了在接下来的主动学习轮次中提供一个预测活性高和不确定性高的分子之间的平衡。然后,如同之前一样,通过相对结合自由能计算确定了这批分子的预测结合自由能,并构建了一套新的3D-QSAR模型。
本轮的共识3D-QSAR模型保持了较高的预测性(Q²值约为0.8,Kendall’s tau值约为0.7),但由于其构建方式,预计相较于第一个模型它将具有更广泛的应用域。虽然可以在“探索”模式下进行多轮主动学习,但我们的意图是评估在非常有限的计算资源假设下,主动学习工作流的效果如何,因此只进行了一次最终的主动学习轮次。
在这一最终轮次中,3D-QSAR模型被应用于Spark结果数据集中剩余的440个分子,并从中选取了排名靠前的20个分子进行自由能计算。因此,在主动学习过程结束时,总共使用自由能计算评估了80个分子,这仅占由Spark识别的生物等排体替换库的16%。
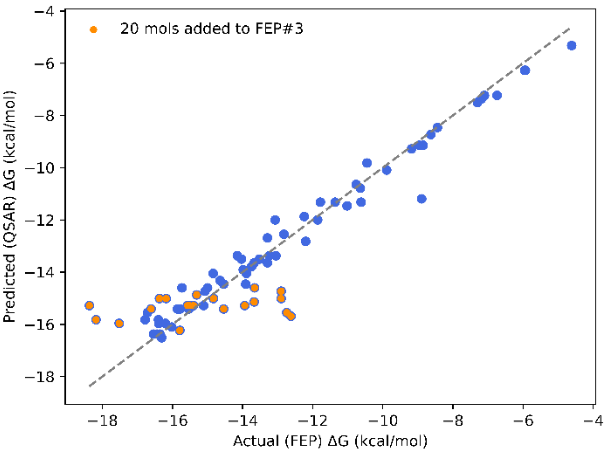
图3. 散点图突显了最终3D-QSAR模型的高预测能力。使用该模型预测靠前的20个分子(被选中进行FEP #3计算)以橙色高亮显示,而用于训练模型的分子则以蓝色显示。
查看结果,主动学习协议在构建预测模型和识别能够提升活性的生物等排体替换方面表现出色。将模型预测与计算得到的结合自由能进行比较显示,该模型在识别紧密结合的生物等排体替换方面极其预测性和有效性(图3)。
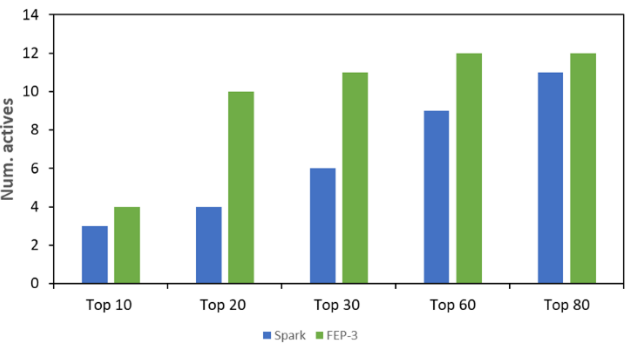
图4. 柱状图展示了从Spark和经过我们主动学习工作流最后一个阶段后检索到的排名靠前分子中所有活性分子的数量。有关工作流不同阶段的详细分解,请参见图SM 4。
在Spark数据集中,大约45%已知活性分子被选中进行自由能计算(富集因子为2.8)。此外,在主动学习工作流的最终排序靠前20名中检索到了10个已知的活性分子,包括位于第3位的Zopolrestat(该分子曾进入临床试验阶段16),而原始的Spark排序仅检索到了4个(图4)。
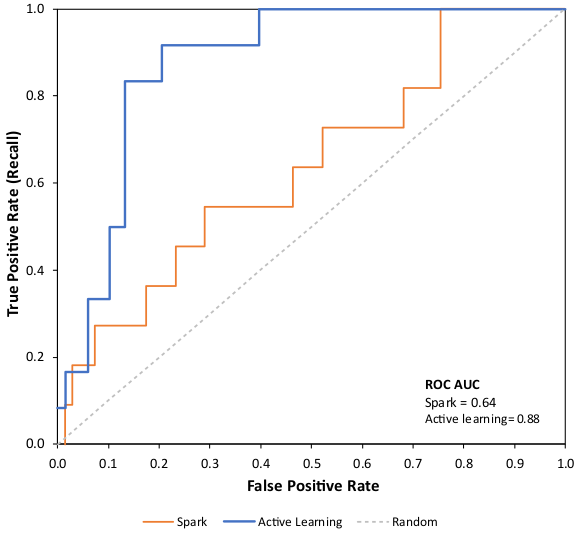
图5. 考虑前80个结果的ROC曲线下面积,突显了我们主动学习工作流(蓝色)在早期富集活性分子方面的优势,与不使用任何优化方法直接从Spark结果池中挑选活性分子(橙色)形成了对比。完整的数据集的ROC AUC请参见图SM 5。
基于Spark和我们主动学习工作流的前80个结果的ROC曲线下的面积(AUC)突显了我们方法性能的提升(ROC AUC=0.88),相较于直接从生物等排体替换实验中挑选活性分子(ROC AUC=0.64),如图5所示。
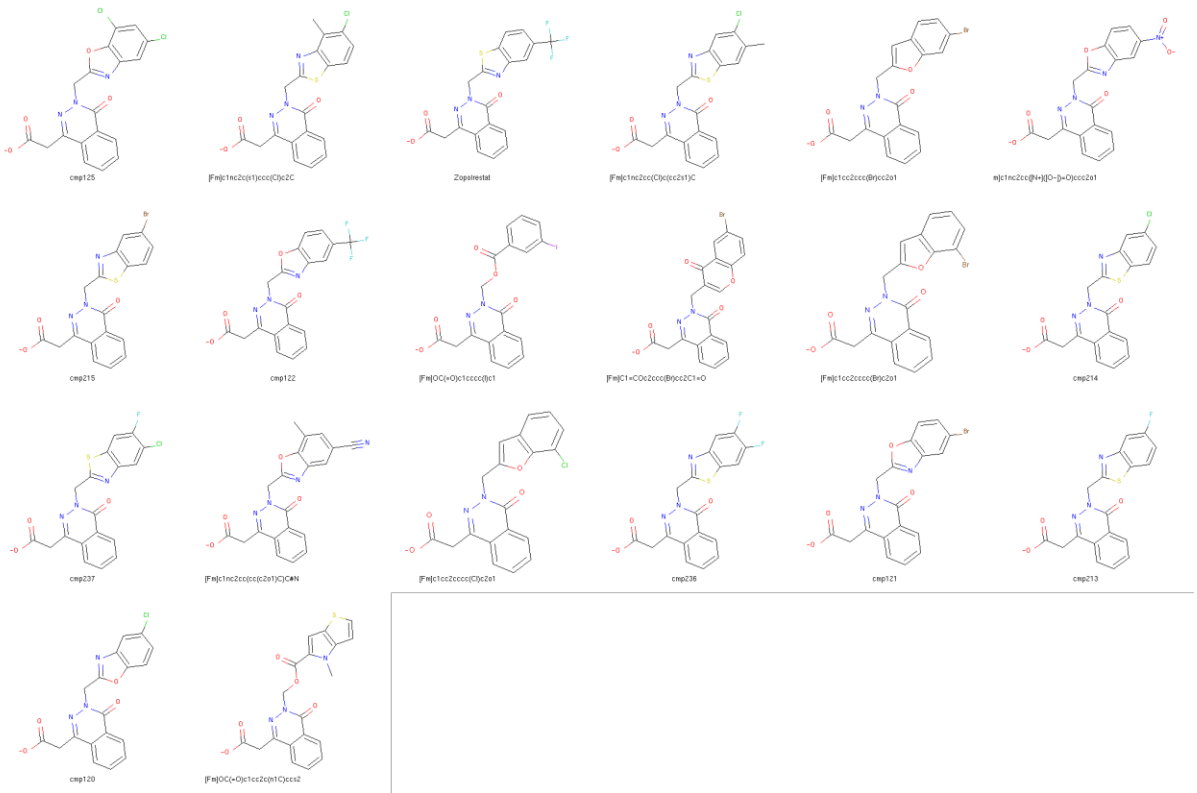
图6. 我们主动学习工作流中排名前20的分子按照其FEP预测的结合亲和力从左上到右下进行排序。已知活性分子根据其化合物名称进行标记15,16,而通过Spark获得的没有文献报告活性的新分子则以[Fm]为前缀进行标记。
从我们的主动学习FEP工作流中优先考虑的前20个化合物中,包含了几种含有苯并噻唑和苯并噁唑侧链的分子,这类化合物系列之前已经被Mylari等人15,16测试过并发现具有活性分子(图6)。在分析与ALR2形成平衡复合物的前5个分子的结合模式时,我们发现它们都与以下两种相互作用有关:(i) 它们的生物等排体替换与疏水特异性口袋中的Trp111之间形成了完美的π-π堆积相互作用;以及(ii) 它们的羧酸头基团与催化阴离子结合位点中的Tyr48、His110和Trp111的侧链之间形成了短接触(氢键),这种结合模式对醛糖还原酶具有高度选择性,而不是对醛还原酶20–22(图7)。此外,由于特异性口袋的高度可塑性——其残基Phe122、Ala299和Leu300能够占据不同的旋转异构体状态,从而以不同方式打开以容纳不同的抑制剂——这为探索Spark建议的那些排名靠前但没有文献报道活性数据的生物等排体提供了路径22–24。
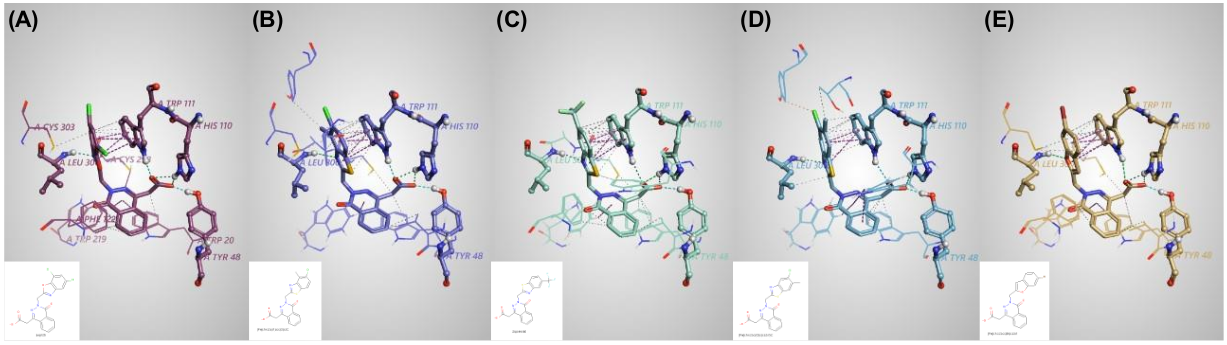
图7. 我们主动学习工作流中排名前5的分子与ALR2蛋白在其平衡复合物中的结合位点相互作用,这些复合物来源于Flare FEP。所有复合物都显示了生物等排体替换与TRP111之间的完美π-π堆积相互作用,以及羧酸头基团在阴离子催化结合位点的短接触(如氢键)。分子按照其排名从左到右排列。(A)和(C)分别是已知的ALR2抑制剂cmp125和Zopolrestat15。
表1. 在Spark结果中排名前20的已知活性分子的Spark排名和pIC50数据
| Spark Rank | pIC50 |
|---|---|
| 2 | 7.51 |
| 4 | 7.11 |
| 8 | 8.51 |
| 16 | 7.59 |
请注意:排名最前的分子是输入给Spark的起始分子
表2. 在我们主动学习工作流结果的前20名中找到的已知活性分子的FEP排名、FEP预测结合能量、报告的pIC50值以及Spark排名
| FEP Rank | FEP ΔG (kcal/mol) | pIC50 | Spark rank |
|---|---|---|---|
| 1 | -18.37(± 1.48) | 7.49 | 63 |
| 3 | -17.52(± 3.15) | 8.51 | 107 |
| 7 | -16.64(± 1.48) | 8.51 | 8 |
| 8 | -16.44(± 1.77) | 7.51 | 26 |
| 12 | -16.36(± 1.45) | 8.15 | 21 |
| 13 | -16.31(± 1.77) | 6.96 | 91 |
| 16 | -16.02(± 1.72) | 7.17 | 349 |
| 17 | -15.86(± 1.46) | 7.57 | 2 |
| 18 | 15.79(± 1.48) | 8.16 | 62 |
| 19 | -15.78(± 1.48) | 7.11 | 4 |
误差值采用bootstrap估算,包括多重Boltzmann平均重新加权(MBAR)误差、滞后性(不同方向进行计算时可能存在的差异)和循环闭合误差(确保计算路径形成闭合环路以验证能量一致性)在内。
总体而言,我们的主动学习工作流展示了如何通过3D-QSAR的快速预测能力和Flare FEP严谨的亲和力预测及排序之间的协同应用,显著提高从数百个生物等排体中选择潜在先导化合物(活性分子)的成功率,同时仅需原本所需计算成本的一小部分(本工作流使用的计算时间不到计算所有500个Spark结果FEP数据所需时间的20%)。在从Cresset Spark建议的500个先导样的化合物中优选ALR2抑制剂的情况下,我们的工作流将成功率提高了近2.5倍。发现只需最少的主动学习循环次数即可获得合理的改进,而不会过多消耗计算资源。我们相信,我们的主动学习工作流具有更广泛的应用前景,并可以应用于其他适合3D-QSAR和FEP的体系。
方法
生物等排体替换实验
搜索ChEMBL数据库25找到由同一研究团队报告的人类ALR2抑制剂及其活性数据(IC50)。将cmp126(pIC50=5.21)选为苗头到先导优化工作流中的苗头化合物。将人类ALR2与Zopolrestat复合物的X-衍射结构(PDB: 2FZ826)导入到Flare V8.027中,经过准备后作为起点使用。在Flare中,使用基于子结构叠合的方法将cmp126叠合到共结晶配体Zopolrestat上。叠合后的化合物(cmp126)随后用作Cresset Spark2进行R-基团替换的起始分子。准备好的蛋白结构以保留输入的质子化状态,移除了所有晶体水的,并作为排除体积导入Spark中,用于指导搜索。选定起始分子中的苯并噁唑部分为要替换的区域(图2-B)。使用默认设置搜索ChEMBL_common和Commercial Common and Very Common数据库。这些Spark片段数据库目前包含了95,568个用于R-基团替换实验的片段,这些片段来源于文献(ChEMBL数据库)(https://www.ebi.ac.uk/chembl) 和商业化合物(VeryCommon和Common)(https://www.emolecules.com/info/products-screening-compounds) 中报道的化合物。
使用上述详述的Spark设置进行的R-基团替换实验生成了500个建议,这些建议基于50%的场相似性和50%的形状相似性,生物等排体因子(BIF%)值范围从56到82。BIF%是一个缩放后的打分,其中原始分子打分为100%,而删除要替换部分的打分为零。正值表示有利的生物等排体,负值则反映一个可以重现原始分子几何结构但对被删除部分模仿效果较差的替换。
自由能微扰计算
自由能计算使用Flare V8.0的标准协议进行。人类ALR2的晶体结构(PDB: 2FZ8)通过Flare进行了准备,并使用TSAR算法28归属了质子化状态。配体的质子化状态保留了从Spark获得的结果,没有进行任何后处理。蛋白质使用AMBER FF14SB29力场描述,配体则使用OpenFF 2.1.030(https://zenodo.org/records/7889050),并采用AM1-BCC电荷。对所有手性、旋转异构体和互变异构状态进行了检查,并在需要时将其包含在FEP图中。体系使用TIP3P水模型溶剂化,在截断的八面体溶剂化盒中,溶剂盒缓冲区为12 Å。仅在平衡阶段使用GCNCMC31采样协议,采样球的缓冲区大小为4 Å。根据需要添加Na+和Cl-离子以中和体系。涉及氢原子的键使用SHAKE算法约束,进行了氢质量重分布,并在整个模拟过程中使用了4fs的时间步长。长程静电相互作用使用粒子网格Ewald(PME)方法处理,非键截断距离为9 Å。体系使用OpenMM包32按照默认协议在λ=0条件下进行平衡。
围绕网络是使用Flare中实现的定制版LOMAP33创建的,当链接打分(link score)低于0.4的阈值时,尽可能自动添加中间状态。此外,在生产模式(production mode)下,从任何配体到已知活性分子(cmp126)的最大距离被设置为3。每个微扰都在正向和反向两个独立的模拟中进行。每个转换的λ窗口数量通过自适应λ窗口调度34自动评估。每个窗口进行了4纳秒的分子动力学(MD)模拟,但对于表现出较大滞后性(hysteresis)的更具挑战性的微扰,则使用了额外的λ窗口和更长时间的模拟。失败的链接或具有大滞后的链接会被重新计算。静电和范德华相互作用使用软核势描述。描述微扰的输入文件使用BioSimSpace35生成的,模拟则是通过SOMD19执行的。自由能及其误差通过MBAR36计算。每对配体结合自由能变化的最终结果是通过平均双向微扰的结果得到。ΔG值根据Xu的方法37从网络中计算得出。
Flare FEP在ALR2数据集上的性能通过一组从已知活性化合物15,16中随机选择的10个化合物进行了评估。我们的协议达到了决定系数(R²)为0.8和平均无符号误差(MUE)为1.6 kcal/mol,这表明了其在本案例研究中的适用性和可靠性(图SM 6)。作为主动学习工作流的一部分,FEP总共对109个配体(包括中间体)进行了计算,涉及249个链接和499个微扰。
QSAR模型的建立
基于FEP衍生的结合自由能计算以及Cresset的三维形状和静电描述符构建了共识回归模型和高斯过程回归模型3。共识回归模型由支持向量机回归(SVM Regression)、随机森林回归(Random Forest Regression)、多层感知器回归(MLP Regression)和高斯过程回归组成。使用k=5的k折交叉验证评估了这些模型的预测性能。
支持信息
全部的500个生物等排体替换的结果及其FEP和QSAR预测的活性值(SDF格式): Molecular structure file。
附加的分析图表和生物等排体替换结果中已知分子的二维表示:Supplementary material。
文献
忽略,请参见原文。
联系我们
获取免费试用版软件在自己的项目中亲自使用3D-QSAR主动学习FEP,或者请联系我们进行商务合作:info@molcalx.com。
原创文章,作者:小墨,如若转载,请注明出处:《3D-QSAR与FEP联用主动学习对药物化学生物等排体进行优先级排序》http://blog.molcalx.com.cn/2025/01/05/active-learning-fep-using-3d-qsar.html


