
摘要:Flare V10发布,新版Flare为计算化学和药物化学带来了新增和增强的科学功能。包括显著改进的相对FEP计算、新增绝对FEP计算、蛋白-蛋白对接以及蛋白-配体相互作用指纹(PLIF)。还通过引入梯度提升和到模型距离度量,扩展并增强了已有的QSAR模型。此外,新增的“Match 3D“可以根据蛋白质的二级结构进行叠合,新增“PSA”按钮可以计算配体的极性表面积等等。
新版Flare为计算化学家和药物化学家带来了新的和增强的科学功能。这些功能包括显著改进的FEP计算(包括绝对FEP)、蛋白-蛋白对接以及蛋白-配体相互作用指纹(PLIF)。
在这个版本中,我们还通过引入梯度提升和模型距离度量,扩展并增强了已有的QSAR模型。此外,新增的“Match 3D“功能可以根据蛋白质的二级结构进行叠合,新增“PSA”按钮可以计算配体的极性表面积。
图1. Flare V10为每一位计算化学和药物化学专家提供了一系列新的功能
用绝对FEP计算精确地预测配体的结合亲和力
绝对结合自由能(Absolute Binding Free Energy,ABFE)微扰计算提供了一种稳健的方法来准确预测结合亲和力。与需要一系列同系物分子进行比较的相对自由能微扰(Relative Free Energy Perturbation,RFEP)不同,ABFE可以独立预测每个分子的结合亲和力。这使得ABFE成为早期药物发现中不可或缺的工具,特别是在苗头化合物识别阶段,能够评估一系列化合物的结合亲和力,为潜在候选药物的后续开发提供关键性见解。
Flare绝对FEP(Absolute FEP,AB-FEP)的实现利用SOMD1作为引擎进行快速的、GPU加速的计算,同时保持用户友好的图形用户界面,确保易于提交计算。
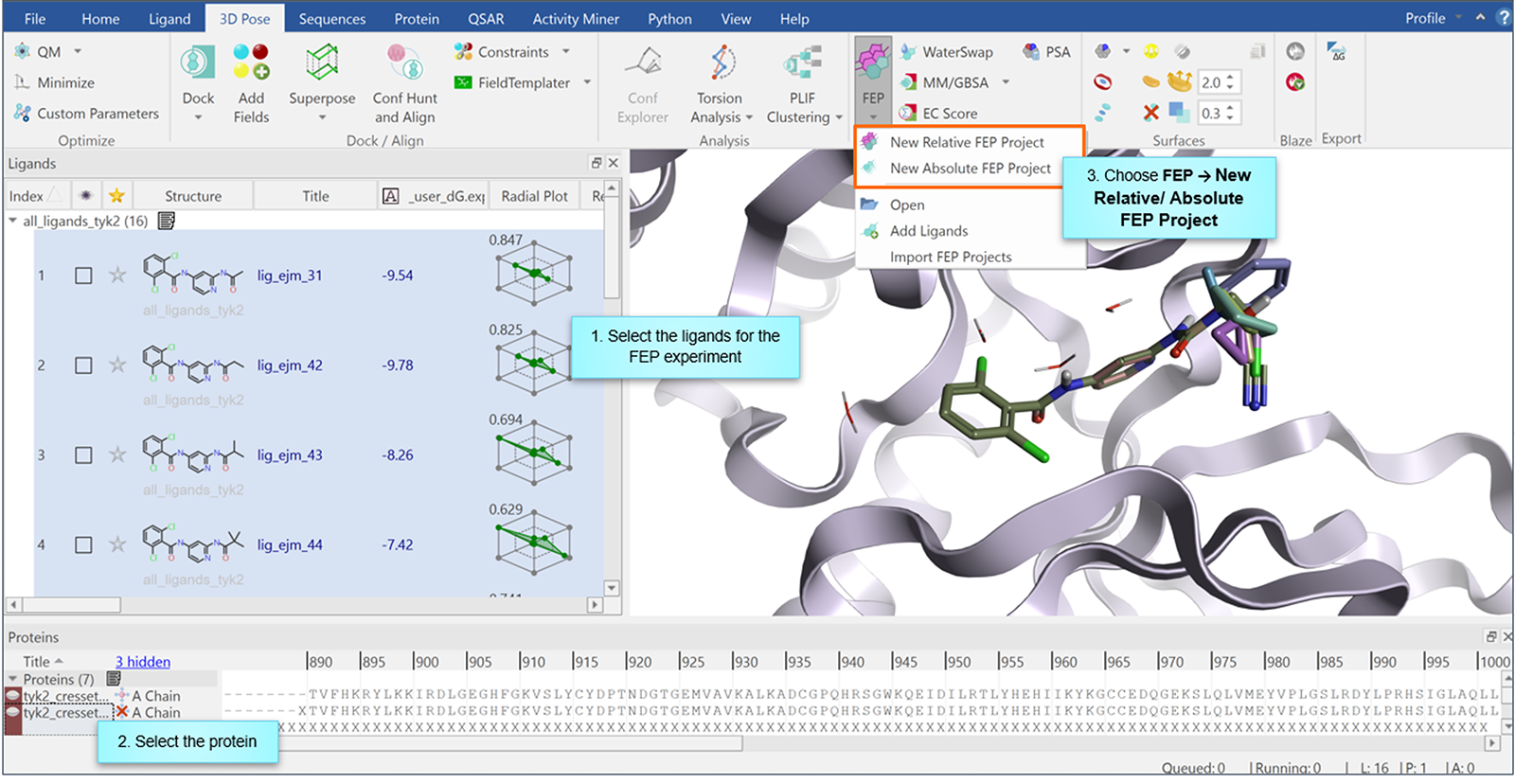
图2. Flare用户友好的界面允许您通过点击三个按钮开始绝对FEP实验
它还允许在单个微扰图中无缝把绝对和相对FEP实验合并,这在引入结构明显不同配体的情况下特别有用。此时,ABFE计算可以在一次运行中直接估算配体的ΔG值,消除了与相对FEP通常涉及显著结构差异场景中遇到的挑战。
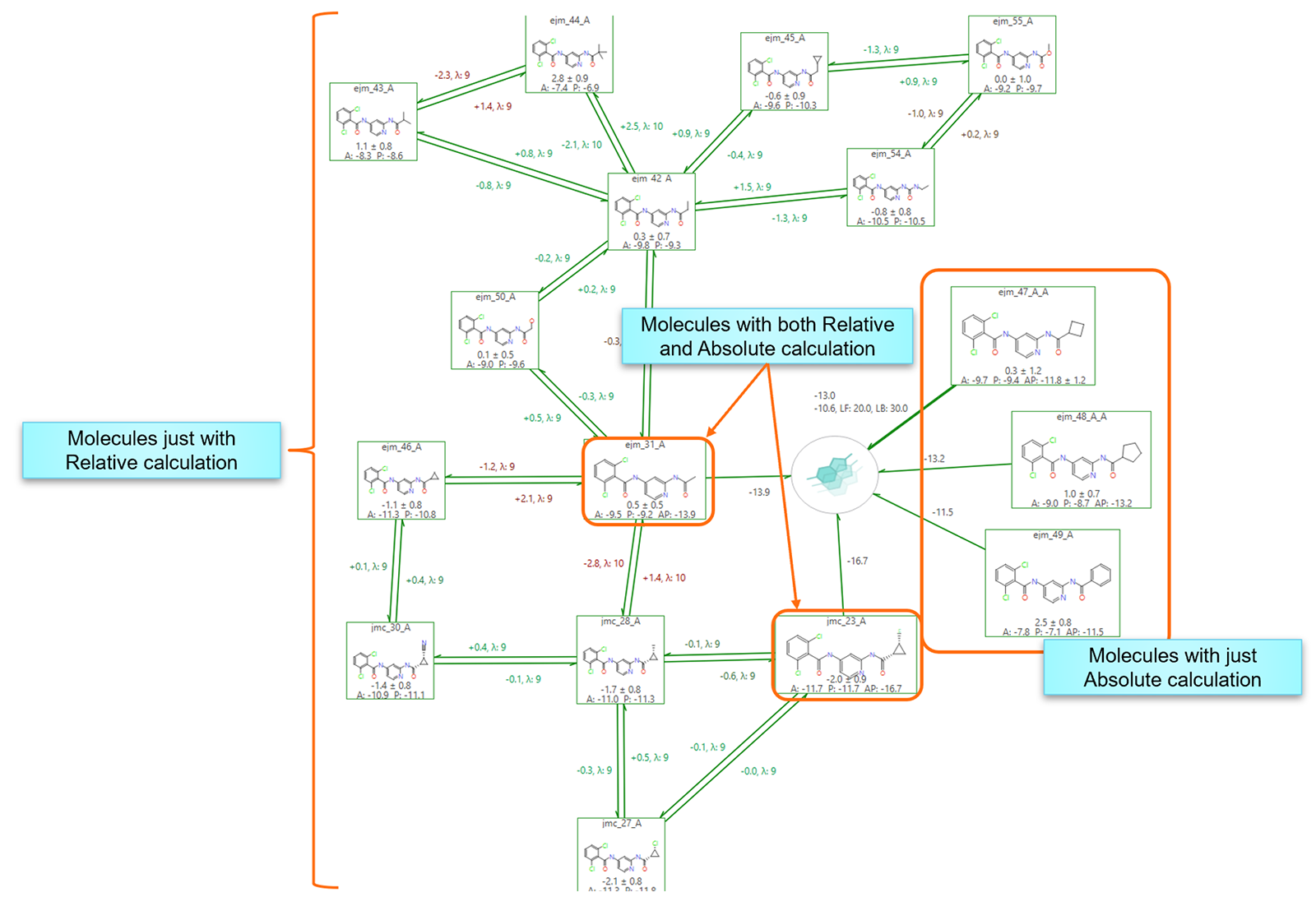
图3. 在Flare中合并完成计算的相对和绝对FEP图
我们的方法已经在不同的场景中进行了广泛测试,并在各种分子体系中展示了准确度、鲁棒性和可靠性,使其成为预测结合亲和力和分子相互作用的值得信赖的方法。
用蛋白-配体相互作用指纹(PLIF)根据配体与蛋白质的相互作用特征对配体进行分析和聚类
蛋白-配体相互作用指纹(Protein-Ligand Interaction Fingerprint,PLIF)是一种用于表示和分析蛋白质与配体之间相互作用的计算方法。PLIF捕获关键的分子间相互作用,如氢键、疏水接触、盐桥,并将其编码为数字指纹。指纹中的每个元素对应于结合位点中特定残基的一种特定类型的相互作用。
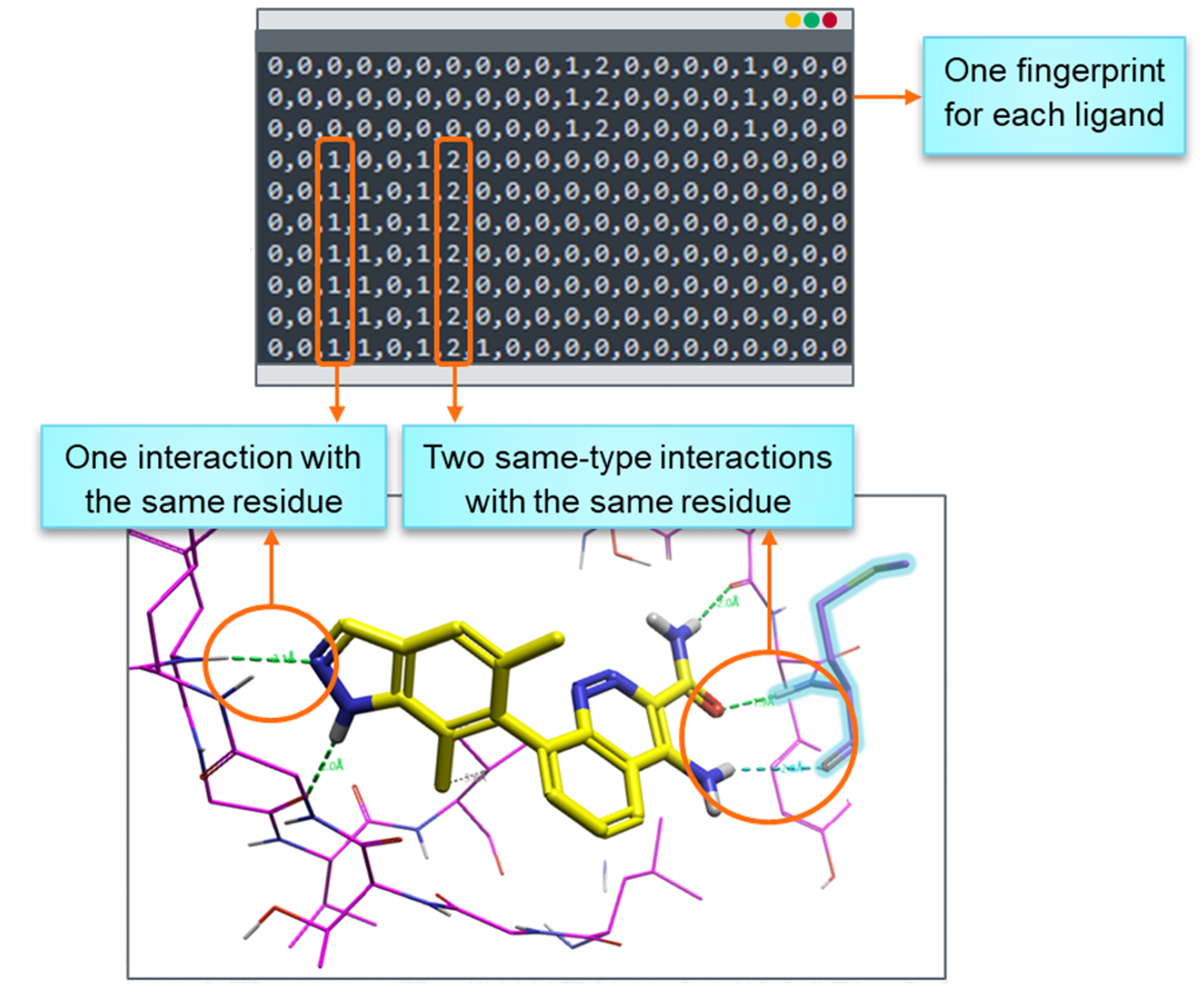
图4. 相互作用指纹可视化有助于识别每个残基的相互作用次数
在新版本的Flare中实现的PLIF通过聚类使得根据配体与靶蛋白相互作用谱进行快速比较成为可能。它还能够识别与强结合亲和力相关的关键相互作用模式,有助于对配体进行优先级排序,优选出具有这些关键相互作用的配体以进一步优化。
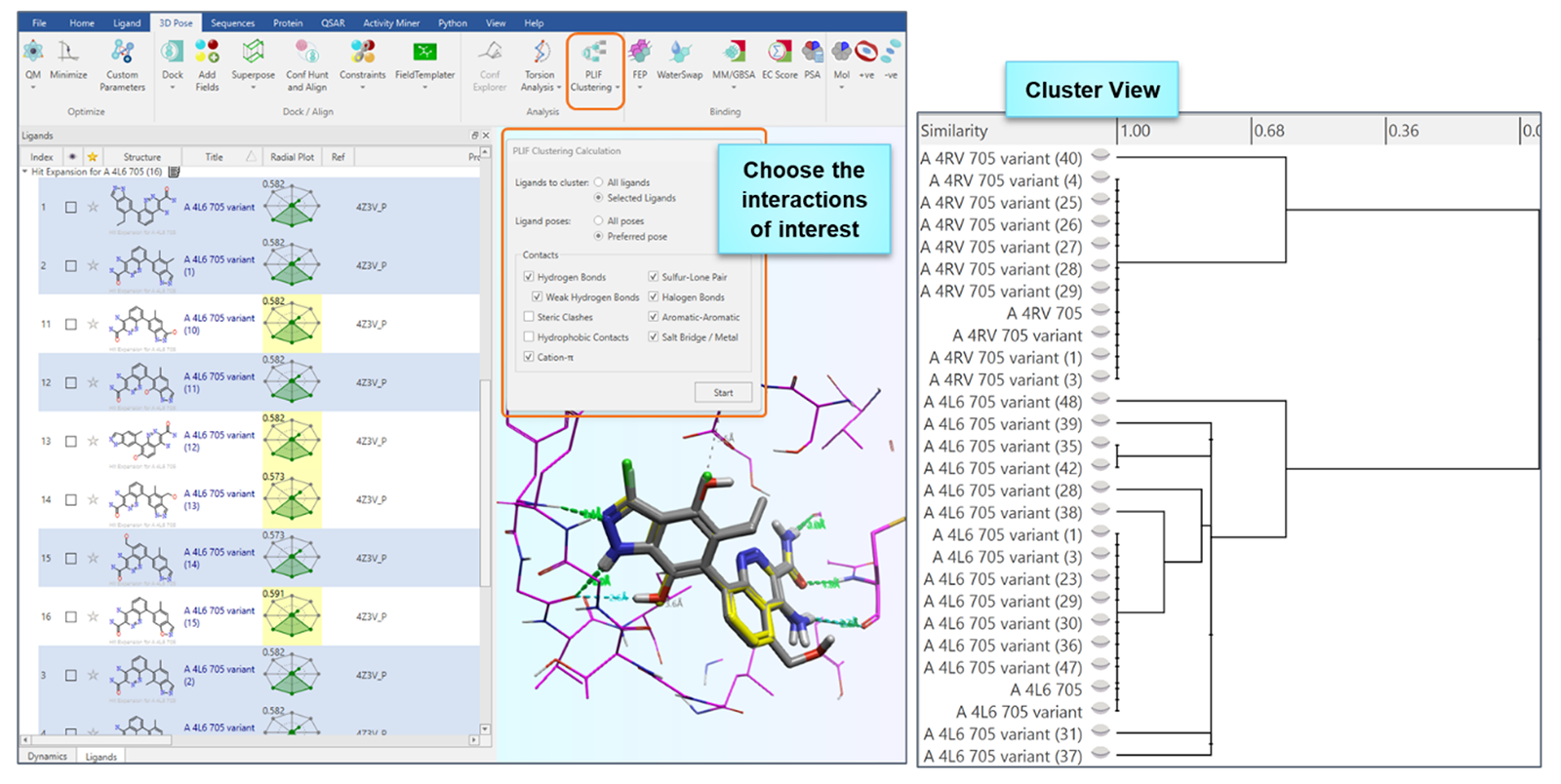
图5. 在PLIF聚类实验之后,使用PLIF指纹相似度对分子进行聚类,并通过树状图在PLIF聚类视图中显示结果。
在Flare中用新增的蛋白-蛋白对接预测蛋白-蛋白相互作用与结合
蛋白-蛋白对接是一种用于预测两个蛋白如何相互作用并结合形成稳定复合物的计算技术,侧重于根据它们的各自结构来识别最可能的结合取向和相互作用界面。它可以应用于预测蛋白-蛋白复合物结构、蛋白二聚体、蛋白-肽复合物以及多聚体组装,后者是通过迭代对接工作流实现的。
在Flare中,这个过程利用了JabberDock算法2,该算法采用粒子群优化(Particle Swarm Optimization,PSO)来探索结合伴侣之间的表面互补性。该方法使用从蛋白质动力学轨迹衍生的空间和时间影响密度(Spatial and Temporal Influence Density,STID)图,以捕捉其形状、静电和局部动态,从而实现精确的对接预测。
要在Flare中运行蛋白-蛋白对接实验,感兴趣的蛋白应与一个短的分子动力学模拟轨迹相关联。该轨迹用于计算STID图。通常,较大的生物分子被指定为“受体”,而较小的则作为“配体”。在实验结束后,“受体”蛋白以及“配体”蛋白打分最高的结合模式将保存在蛋白质表单中。
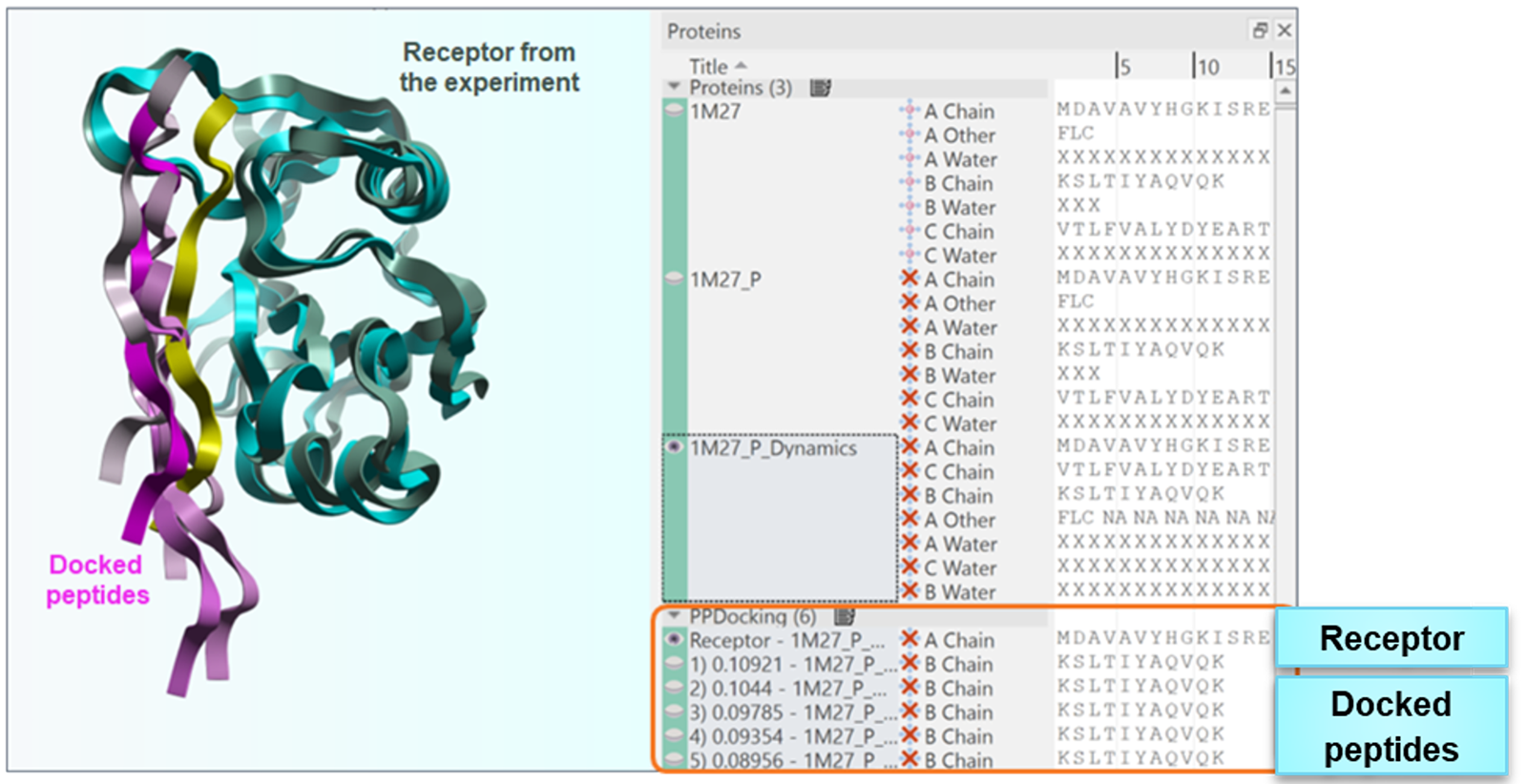
图6. 蛋白质-蛋白质对接实验成功地识别出了大部分的正确结合模式
梯度提升(Gradient Boosting)和到模型的距离(Distance to Model)指标增强的QSAR模型
在Flare V10中新引入梯度提升功能和到模型的距离指标,扩展并优化了其QSAR能力。
梯度提升(Gradient Boosting):
- 捕捉分子特征和生物学活动之间的复杂、非线性关系
- 在保持对过拟合鲁棒性的同时,提供高预测精度
- 高效处理各种数据集,使其成为优化先导化合物和精确预测药物活性或毒性的理想选择
- 使用超参数优化来提高模型性能
- 对于超过1500个分子的数据集,使用直方图梯度提升,在训练前将输入特征分组为整数值的区间,从而提高计算效率,实现更快的处理速度
到模型的距离(Distance to Model):
- 根据感兴趣分子与训练集分子在描述符空间上的距离划分为“内部”、“接近”或“外部”来评估预测的可靠性
- 提供了一个清晰的评估方法来衡量预测分子与训练集数据之间的叠合程度,从而提高了QSAR预测的信心
这些增强功能使得Flare QSAR建模在药物发现的决策中更加灵活、准确和信息丰富。
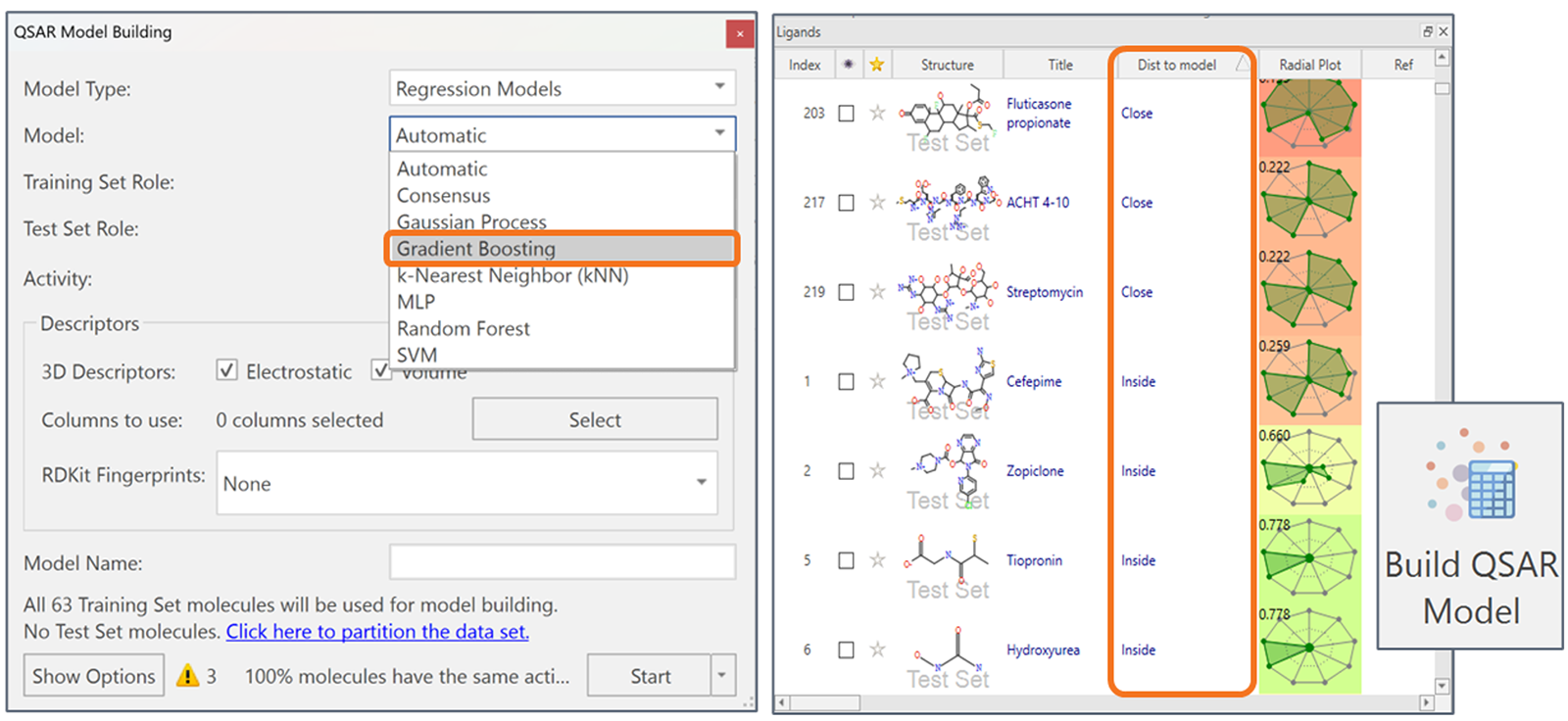
图7. 梯度提升既可用于回归模型,也可用于分类模型。在配体表单中捕获的“Distance to Model”指标用来评估感兴趣的分子是否匹配训练集分子描述符空间的“内部”,“接近”或“外部”。
Match3D:匹配核心二级结构元素对蛋白进行叠合
传统的蛋白叠合方法主要依赖于序列相似性,根据氨基酸序列的相似性对蛋白质进行比对。然而,具有相似功能的蛋白质通常具有共同的关键3D结构特征,并不一定保持高的序列相似性。基于二级结构的叠合可以实现此类区域的比对,即使序列相似性较低。
在Flare中,新增的蛋白质二级结构叠合称为Match3D,它利用通用结构(Universal Structure,US)比对算法3,该算法优化了TM打分,这是一种评估两个蛋白质之间结构相似性的度量标准,独立于它们的序列。同时,Flare现有的“Superpose”功能并不根据二级结构相似性对蛋白质进行叠合,而是通过匹配其序列并最小化对应残基之间的距离来进行叠合,不考虑蛋白质的3D特征。
两种方法之间的差异在下面的例子中得到体现,其中两个蛋白质(pdb代码:1F51和1Y4T)具有相似的结构,但序列相似性低,用两种方法进行叠合。在本算例中,Superpose的结果是一个叠合不好的体系,考虑到低的序列一致性,这是预料之内的。Match3D根据链的3D相似性重新计算最佳等效残基来解决这个问题,从而产生更准确的结构叠合。
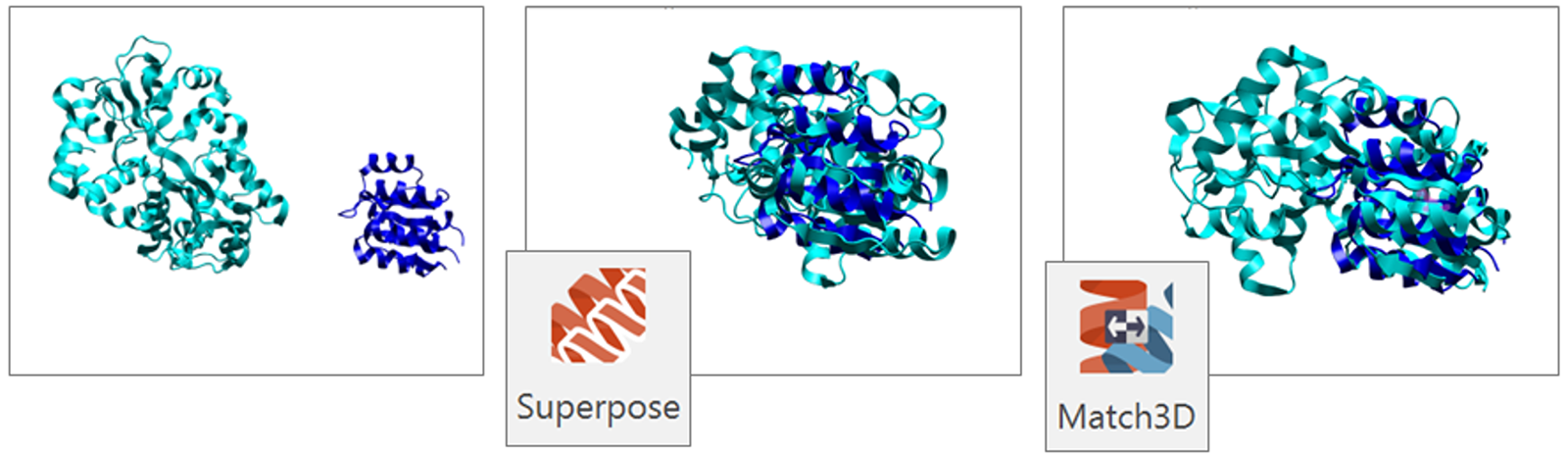
图8. 在蛋白质序列相似性低的情况下,使用Match3D实现了增强的蛋白质叠合
通过关注二级结构,Match3D 提供了更健壮和功能相关的蛋白质比较,解决了基于序列方法的局限性。
Flare V10隐藏的宝藏
无论您是药物化学还是计算化学专家,Flare V10都能为您提供了几个额外的工具,显著增强您的工作。新增的用于计算极性表面积(PSA)和表面面积(SA)的按钮可以帮助您深入了解感兴趣的分子的吸收和渗透能力。除此之外,它还允许您计算与尺寸相关的无缩放静电互补性(Electrostatic Complementarity™,EC)4 打分(\(Unscaled\ EC = EC \times surface\ area\))。该替代打分提供了有助于分析不同大小配体的信息。
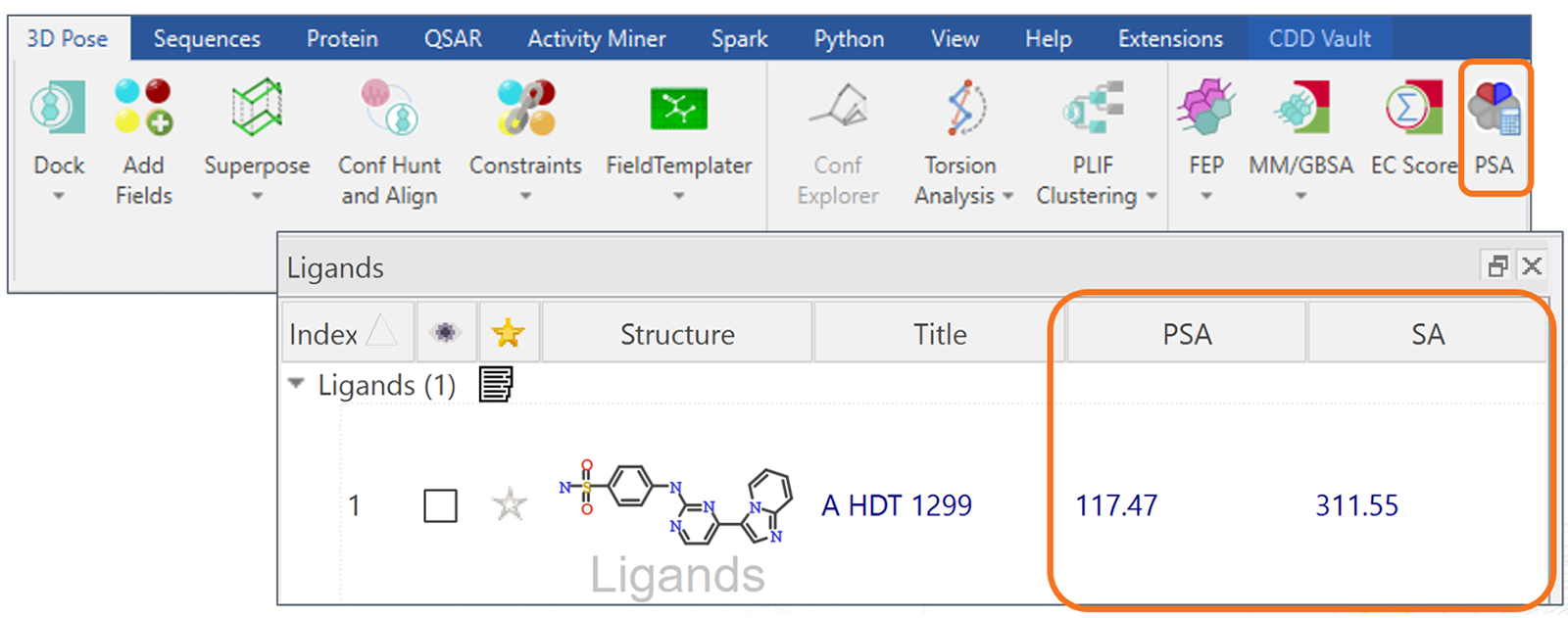
图9. 只需点击一个按钮,即可计算感兴趣配体的极表面积(PSA)和表面面积(SA)。
现在,Flare为那些已知“模板”配体结合模式并希望利用此信息来偏置同系物共价对接结果提供了实用解决方案。通过新增的模板共价对接,要对接的分子将根据子结构与模板配体叠合,并使用叠合后的构象作为对接计算的种子,通常会带来更好的对接结果。
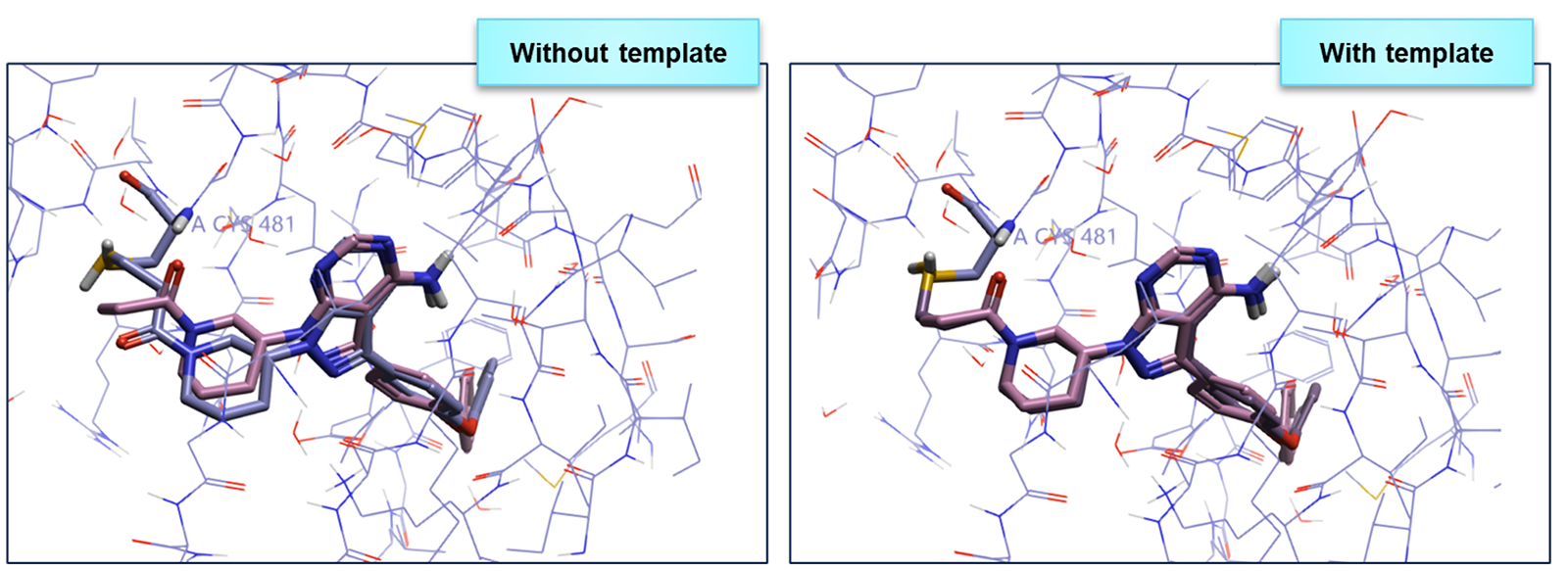
图10. 通过选择一个模板配体来指导共价对接过程,您会得到更好的结果。
此外,Flare V10 引入了增强的量子力学(QM)功能,提供了精确分子分析的工具。现在可以在指定的二面角范围内执行扭转角扫描,并完全控制定义最小和最大角度。结构可以使用 xTB5 在溶液中进行最小化处理,确保在真实条件下几何形状得到优化,同时保持几何优化与单点计算之间的一致性。最后,将碳负离子用作 QM 计算输入的能力扩大了可分析化学体系的范围,在研究分子构象和能量方面提供更大的灵活性和准确性。
Spark™ in Flare:新增与增强的功能
Spark™ in Flare引入了几项增强功能,简化了工作流并提高了易用性。现在,在启动所需的Spark向导之前,您可以在Flare的三维窗口中直接选择配体原子来预先选择要替换的起始分子的R-基团。这项增强功能消除了额外的步骤,节省了时间,并允许更直观地设置。一个新的高级选项允许您为每个分子设置对接时限,从而更好地控制计算效率。此外,Spark结果现在会自动根据实验名称进行标记,以便更好地组织和跟踪结果。
这些增强功能有助于将生物等排体替换研究纳入Flare工作流,并使用户能够以更高的精度、效率和信心进行操作。
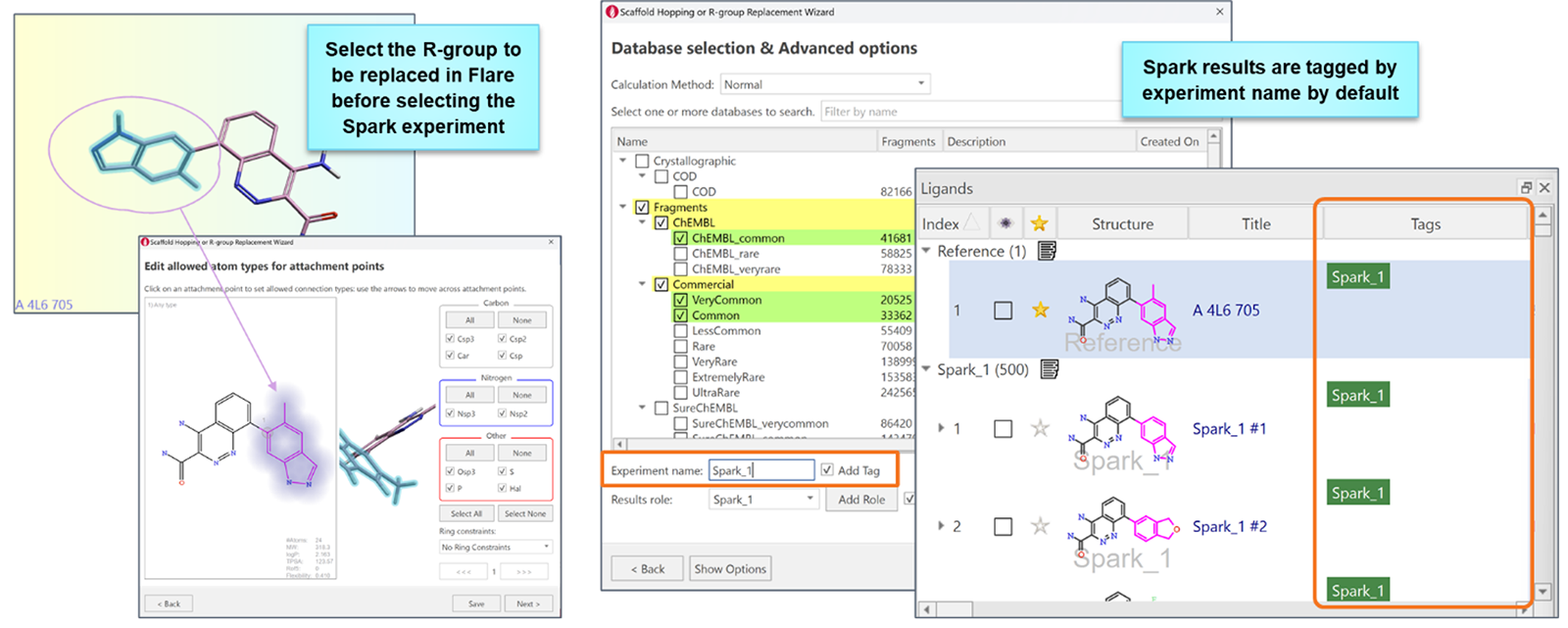
图11. 左:跳过Spark向导步骤,在Flare 3D窗口中选择要替换的分子片段。右:现在默认情况下,Spark结果会自动添加用户提供名称的tag标签。
Flare FEP:新增与增强的功能
V10中的Flare FEP具有多项改进,其中一些如下所示:
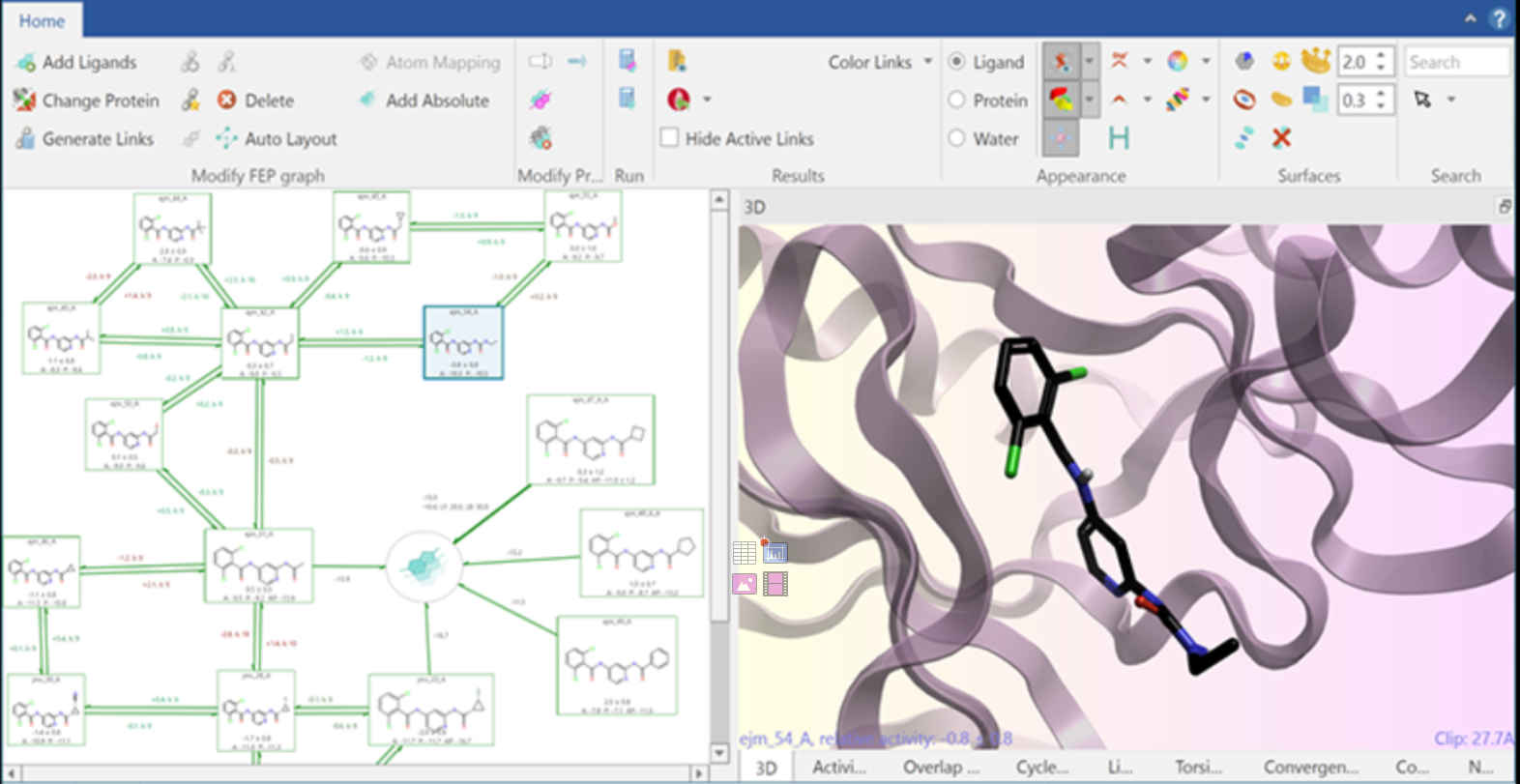
如果您的项目中相对FEP图的分子簇没有很好地连接,您可以使用新增的“Enhance Connectivity”按钮来改善FEP图的连接性。每次点击都会添加一个新的链接,旨在最小化平均最短路径长度的同时优先考虑打分最高的链接。算法从距离减少选项的前10%中选择最佳链接,并强烈偏向于最高链接分值(link score)。
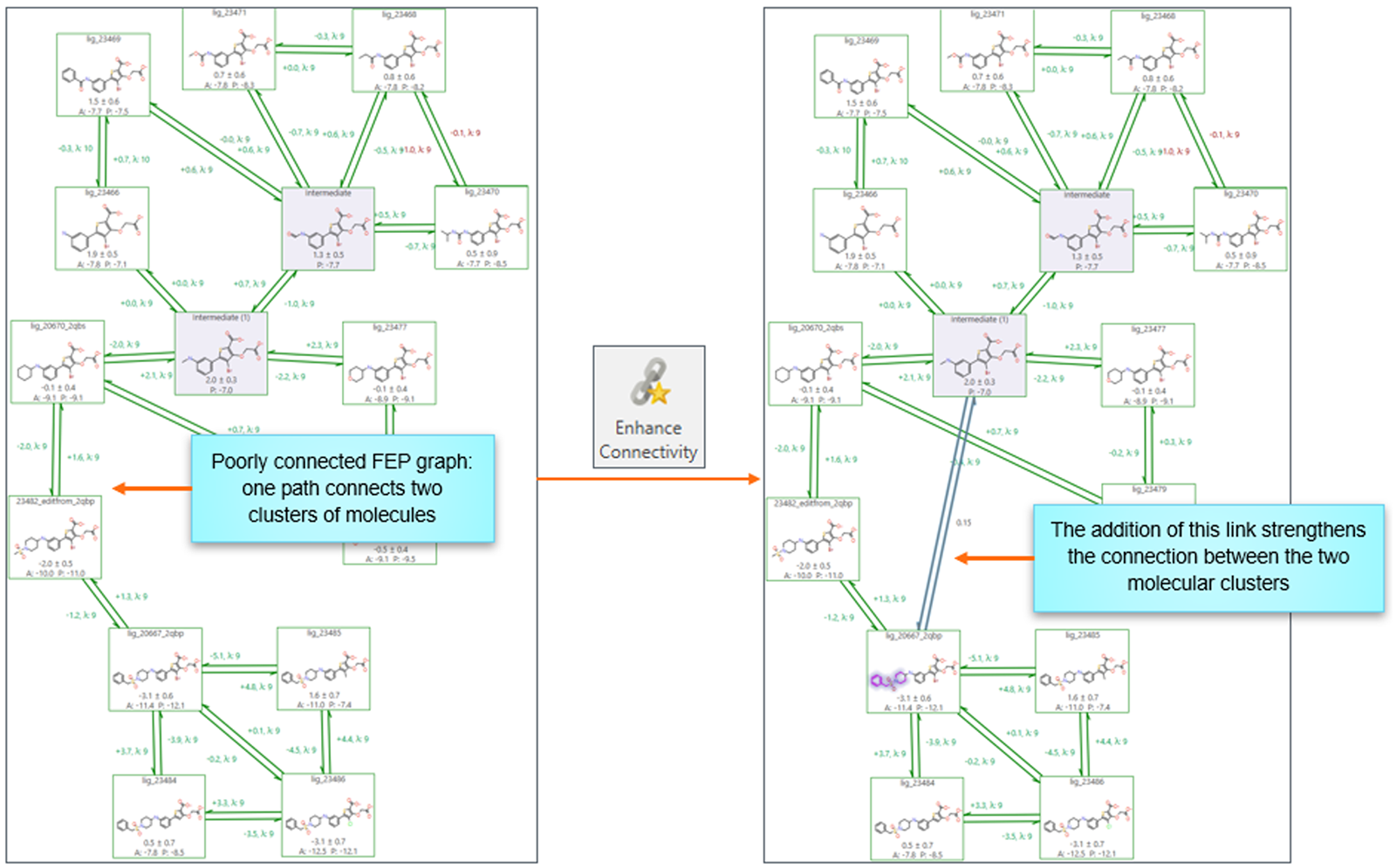
图12. 通过单击按钮添加新链接增强了FEP图中两个聚类簇之间的连接性。
新的“按质量着色(Color links by quality)”会将根据计算出的质量打分为FEP图着色。该打分值考虑了收敛图、重叠矩阵和接触的质量。颜色方案直观:红色表示质量差,灰色表示平均质量,绿色表示高质量。
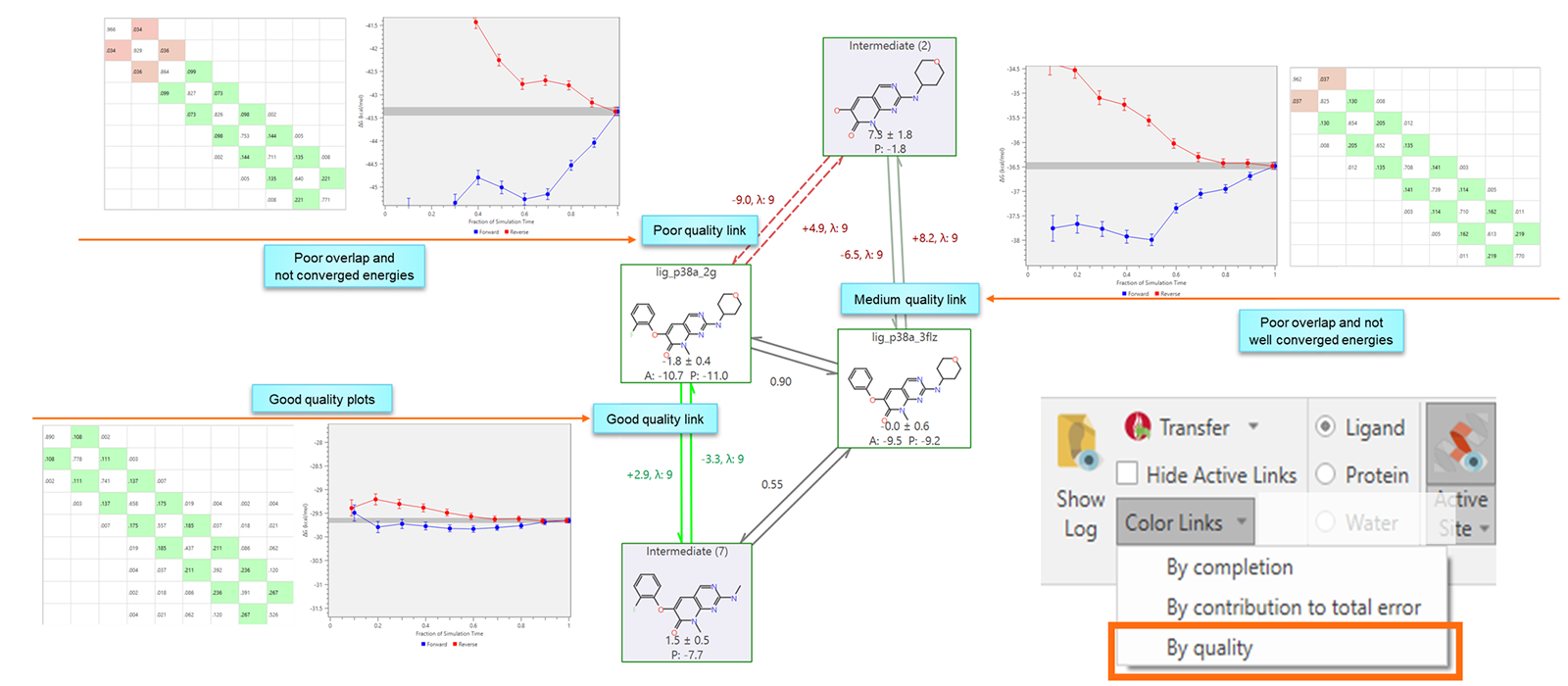
图13. 快速、直观地识别FEP图中有问题的链接。
在增强的活性图中,如果选中“Precision”复选框,则可以查看混淆矩阵(confusion matrix),其中包含所研究配体的真阳性(TP)、真阴性(TN)、假阳性(FP)和假阴性(FN)预测的数量。FEP图的设置也移动到一个新的小部件中,允许您修改轴的范围和单位,并更改图表的背景颜色。
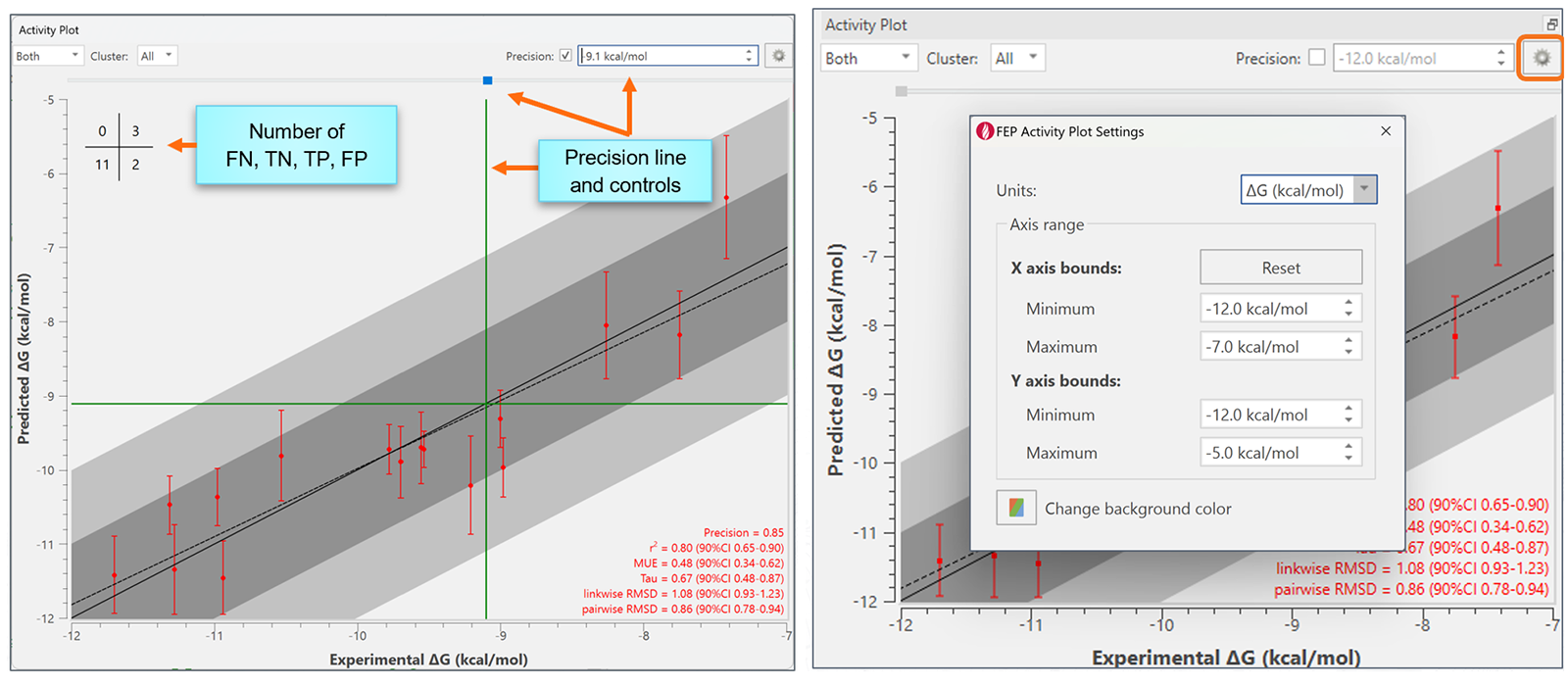
图14. 左:在图表中显示FN、TN、TP和FP的数量。右:活性图的设置现在被分在一个单一的小部件中。
Flare FEP中的其他增强功能包括通过单击“Contact”表单中的行即可在3D视图中突出显示形成接触的原子,向FEP项目日志添加框信息,新的成对(每种化合物)RMSE 统计数据,以及重新启动或继续进行FEP计算时改进了灵活性,允许更改平衡阶段设置并选择打开或关闭GCNCMC。
分子动力学模拟:新增与增强的功能
Flare V10中的Dynamics模块包括分子动力学模拟的几个补充和增强。其中一些如下详细说明:
在Dynamics模块计算过程中,新增的功能可以约束角度、扭转角、距离和位置,允许您模拟具有受控蛋白质主链的动力学。设置距离和位置约束的小部件也已更新以提高用户友好性。所有约束信息也被添加到原子工具提示中。
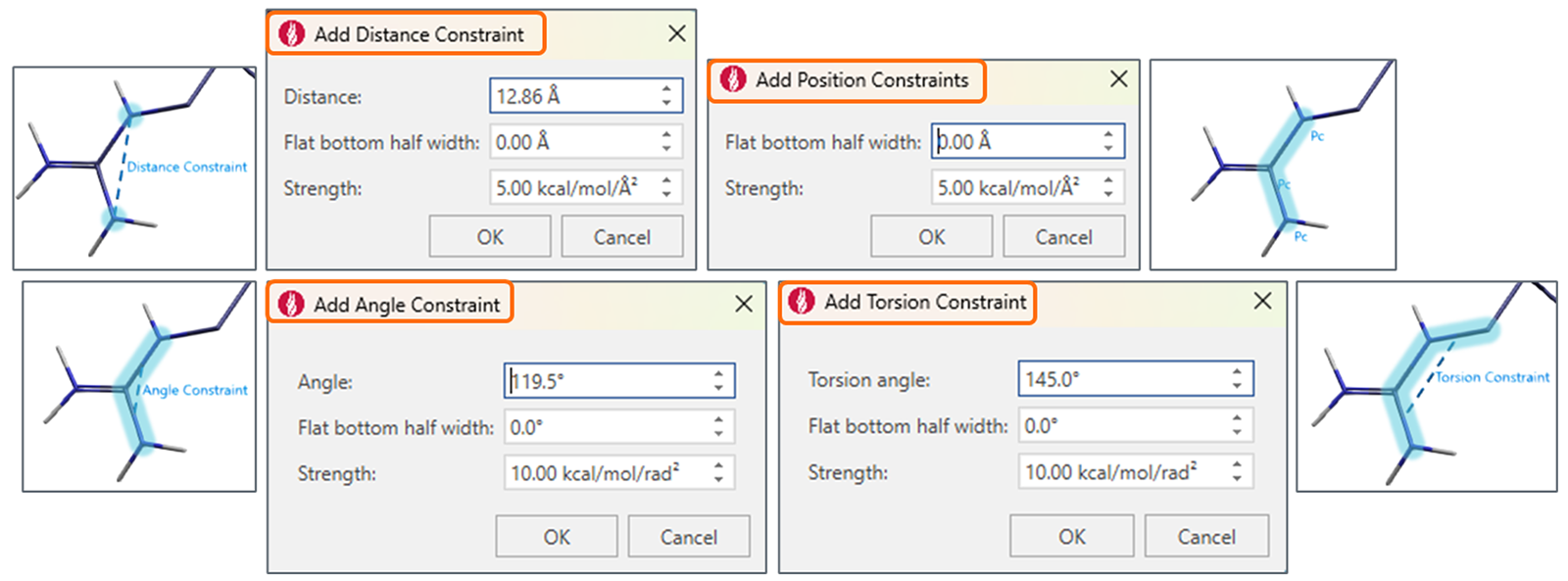
图15. 新增以及增强的小部件用于在动力学模拟过程中对位置、距离、角度和扭转角添加约束
对与轨迹相关的功能进行了几项改进。例如,现在您可以导出不包含溶剂的轨迹,并可以从导入的pdb结构和轨迹文件中恢复分子动力学模拟。如果您希望从轨迹中选择特定帧并保存它,现在的帧复制功能会自动将原始帧的信息附加到新创建的蛋白质上。这确保了所选帧的来源与其源相关联,从而更容易跟踪和管理来自不同部分的动力学轨迹中的蛋白质构象。
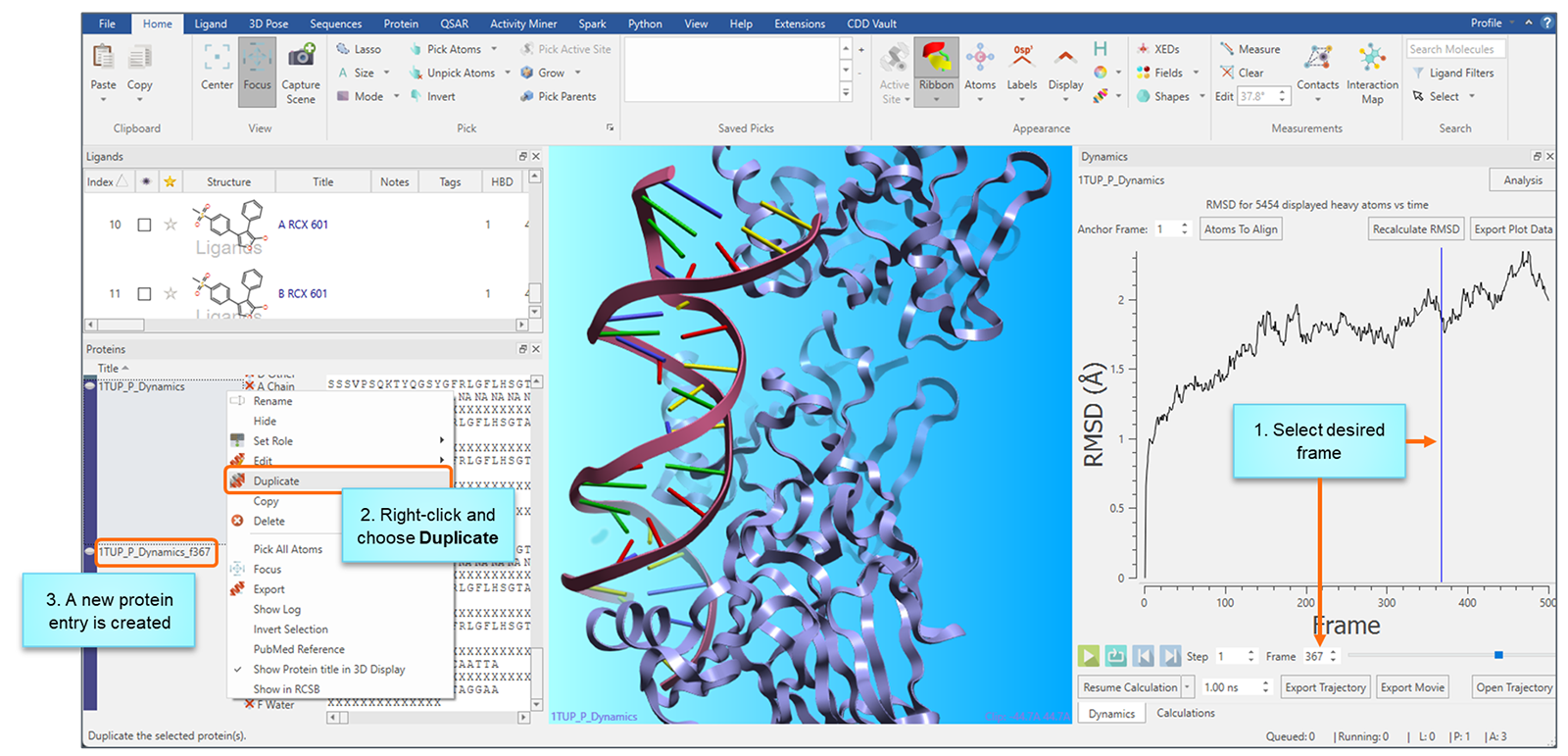
图16. 自动添加帧号到复制的蛋白结构上
其他增强功能,例如在openmmff中添加改进的AMBER 5-磷酸盐参数、能够使用自定义平衡协议导出计算结果、新的按钮可以轻松切换图和识别属于特定主成分分析簇的帧等,这些都只是Flare V10 Dynamics模块中新增功能的一部分。
其它的功能增强与改进
随着每个Flare版本的发布,我们不仅致力于整合令人兴奋的新的科学进步,还致力于提高可用性,这通常基于用户直接反馈。以下是在Flare V10中出现的一些其他值得注意的新功能:
- 如果您正在处理大环化合物或大型分子结构,使用新的增强版最大公共子结构(MCS)基于配体的叠合算法可以显著提高结果
- 新增“HELM”Flare扩展程序,通过HELML表示法创建新肽,并将新的单体/砌块添加到由HELML支持的非天然残基列表中
- 控制3D视窗中显示的蛋白质的透明度
- 创建注释标记球体以映射3D视窗中的空区域。该球体具有可自定义的大小以及可选的关连标签
- 增强Cresset Engine Broker的连接:当计算在多个Broker上启用时,只要可用即可自动启动
- 蛋白准备与结构检查增强了对DNA与RNA的处理
- 增强了FREAD6Loop Modeling模块,在自动模式下将配体、水和辅因子克隆到固定的蛋白质中
在药物设计过程中Flare全程陪你
Flare V10交付先进的科学方法、分析工具和直观、易用的增强功能,洞察您的配体-蛋白质复合物结构。
想要尝试Flare信息丰富、用户友好界面,发现它如何帮助您自信地推动潜在先导化合物优化?请现在就联系我们安排试用,快速访问Flare的广泛功能。我们的专业团队随时准备通过安装和设置为您提供支持,而我们全面的教程库——涵盖从常见工作流程到高级方法和功能的所有内容将帮助您开始使用。我们在这里帮助您更快地实现目标,让您设计出重要的分子。
- 电邮:info@molcalx.com
- 电话:020-38261356
参考文献
- G. Calabro, C. J. Woods, F. Powlesland, A. S. J. S. Mey, A. J. Mulholland, J. Michel, Elucidation of Nonadditive Effects in Protein–Ligand Binding Energies: Thrombin as a Case Study, J. Phys. Chem. B. 2016, 120, 24, 5340–5350
- L.S.P. Rudden, M.T. Degiacomi, Protein Docking Using a Single Representation for Protein Surface, Electrostatics, and Local Dynamics, J. Chem. Theory Comput. 2019, 15, 9, 5135–5143
- C. Zhang, M. Shine, A.M. Pyle, Y. Zhang, US-align: universal structure alignments of proteins, nucleic acids, and macromolecular complexes, Nat. Methods, 2022, 19, 9, 1109-1115
- Bauer M. R., Mackey M. D.; Electrostatic Complementarity as a Fast and Effective Tool to Optimize Binding and Selectivity of Protein–Ligand Complexes, J. Med. Chem. 2019, 62, 6, 3036-3050
- C. Bannwarth, S. Ehlert, S. Grimme, GFN2-xTB—An accurate and broadly parametrized self-consistent tight-binding quantum chemical method with multipole electrostatics and density-dependent dispersion contributions, J. Chem. Theory Comp. 2019, 15, 3, 1652-1671
- Y. Choi and C. M. Deane, FREAD revisited: accurate loop structure prediction using a database search algorithm, Proteins, 2010, 78, 1431-1440


