
计算赋能工作流识别全新KRASG12C共价别构抑制剂
摘要:本文用Ilib Diverse重现了拜尔公司Mortier等人的计算赋能工作流识别全新KRAS G12C共价抑制剂中的组合库设计工作流。总的来说,Ilib Diverse使用非常简便,通过对片段的收集以及自己对项目的理解,可以快速、高效地枚举感兴趣的化合物库,最终实现加速项目进度的目的。虚拟组合库是实现骨架跃迁、先导化合物发现与优化的重要手段,Ilib Diverse让您轻松地实现库的设计。
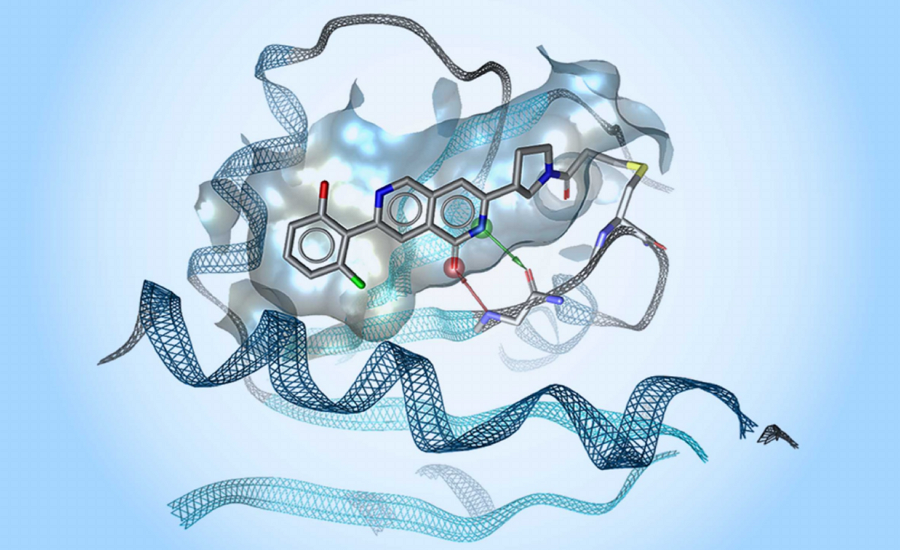
封面说明了一种新型抑制剂以前所未有的结合方式与KRASG12C共价结合(用LigandScout制作的图像)。这一发现是基于计算驱动工作流枚举新化学物质并逐步进行优先级排序的结果。对最有希望的化合物进行了合成、生化测试、并与KRASG12C共结晶。更多信息请阅读Jérémie Mortier等人的原文[1]。
肖高铿/2021-05-24
1. 前言
Bayer公司Mortier等人(2020)[1]发表的ChemMedChem封面文章详细地描述了发现全新KRASG12C共价抑制剂的过程。整个计算工作流如图1所示,作者首先以已知的共价抑制剂ARS-1620(图1 A)为模板分子,将化合物碎片化为4个部分(图1 B):1)丙酰基弹头;2)桥连的哌嗪;3)喹唑啉母核;4)3-氟苯酚头部基团。接着,作者用一个工作流对桥连的哌嗪与母核部分进行骨架跃迁,枚举得到一个含有7×106化合物的数据库,由各种砌块组成,并且这些化合物没有出现在ChEMBL数据库里,这说明这些化合物具有很高的新颖性。其中有5个化合物出现在SureChEMBL里,均来自Araxes的专利[2]。
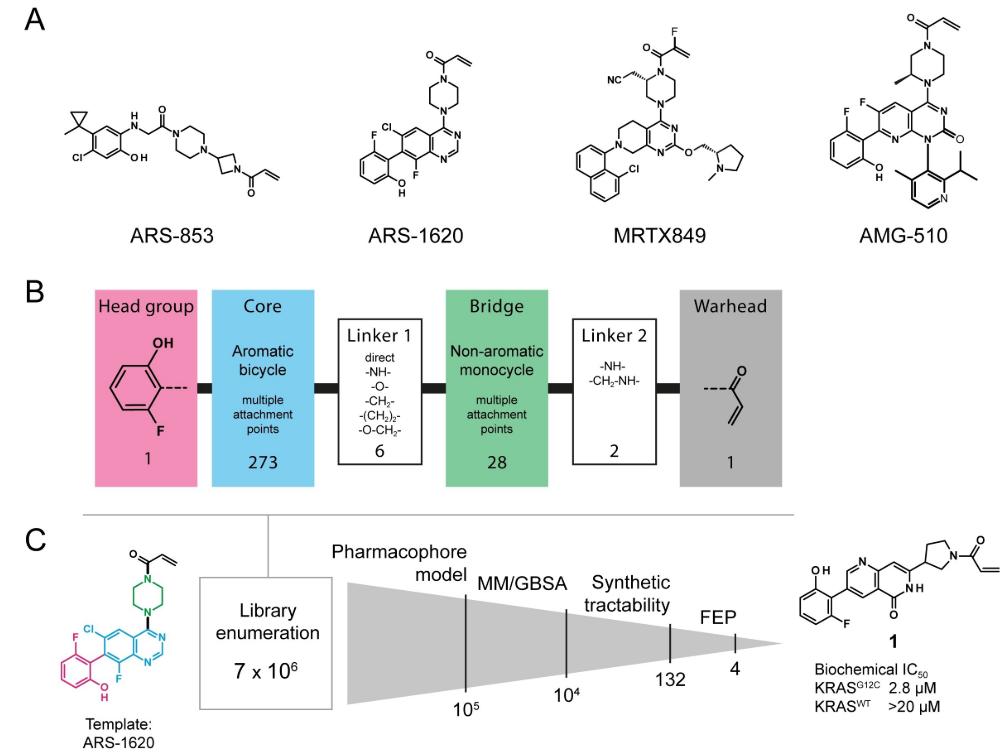
图1. A) 结合到KRASG12C switch-II口袋的已知抑制剂;B)在数据库枚举之前抑制剂结构碎片化策略、碎片库的性质与大小; C)枚举与优先级排序工作流
作者将枚举出的化合物用药效团模型进行虚拟筛选,保留叠合最佳的前105个化合物;接着进行共价对接,并用MM/GBSA打分对化合物进行优先性排序,保留打分最佳的104个化合物;再接着考察化合物的合成可行性,抛弃合成可行性差的化合物,最终保留合成砌块有商品购买的化合物132个;最后,用FEP+方法预测132个化合物的结合亲和力,最终有4个化合物预测的ΔG落在ARS-1620范围内。
在此之前,我们演示了如何实现基于反应的组合库策略进行组合库设计。本文的目的是,补充一种基于片段拼合的策略进行组合库设计。因此,本文重点是实现图1中B的化合物枚举过程。
2. 用Ilib Diverse枚举化合物库重现Bayer建库流程
2.1 规划组合库设计策略
本文以图2所示的简单策略建立虚拟组合库:在该策略里,化合物被划分为1、2、3、4个部分,分别对应Ilib Diverse的4个烧瓶(见图3 flasks部分)。
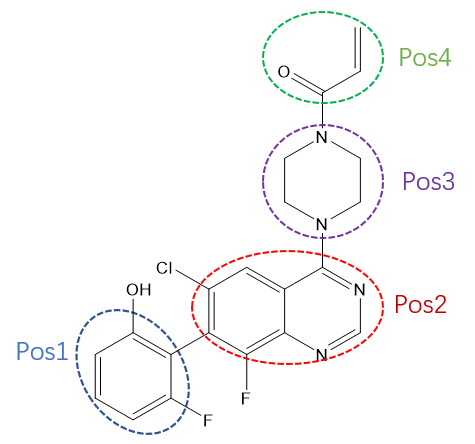
图2. 组合库设计策略
规划建库策略实际就是对模板分子进行碎片化的过程,图2的策略与Mortier等人(2020)[1]的策略是一致的。接下来,我们要定义每个位置需要用什么样的片段来代替。
2.2 用Ilib Diverse枚举化合物库

图3. Ilib Diverse建库的界面与流程
步骤1. 碎片数的设置
根据2.1规划的设计策略,化合物由四个部分组成,因此我们设置碎片数为4,此时Flasks栏出现Pos1、Pos2、Pos3、Pos4四个烧瓶,这四个烧瓶分别对应图2的四个片段顺序。接下来,需要往烧瓶里加入各种片段。
步骤2. 添加碎片到烧瓶
Flask 1:如图3所示,在本算例中,我放置了3个化合物:1,3-二氟苯,3-氟苯酚,喹唑啉。并将1,3-二氟苯与3-氟苯酚的权重(weight)设置为100%,而将喹唑啉的权重设置为25%,这样做的目的是让组合化合物的时候,更多的使用1,3-二氟苯与3-氟苯酚,而少使用喹唑啉。片段的权重可以依据项目需求而设置。再比如,如果是骨架跃迁目的,需要保留ARS-1620的2-氟-5-羟基苯的头部片段(见图1)不变,则Flask 1就仅放置3-氟苯酚。
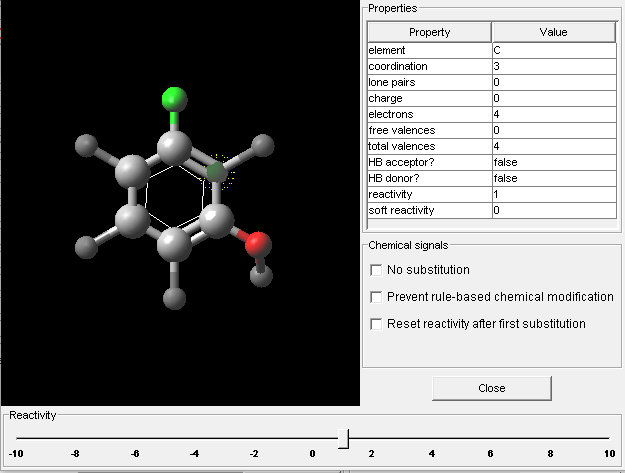
图4. 原子性质编辑器
可以进一步用原子性质编辑器来设置原子的反应性以便控制反应的方向与优先性。以3-氟苯酚为例,如图4所示,设置(1)氟与羟基中间的碳原子的反应性为1,其它碳原子的反应性为默认的0,这么做的目的是控制反应的方向:与flask2片段拼合时,反应性高的原子优先用于拼合,以便组合出的化合物偏向于自己期望的那种。(2)设置酚羟基氧为“No substitution”以确保酚羟基氧不参与后续的反应。
图5. 导入新片段
Ilib Diverse提供了各种基团(见图3的3),点击Group,会列出该基团里收集的各种片段,可以将整个group添加到烧瓶里,也可以选择部分片段添加到烧瓶里。如果,你感兴趣的片段没有列在现有的group里,你可以自己添加:用ChemDraw或其它软件画好结构,保存为mol格式文件,通过菜单Fragment | import new fragment into current group导入到当前Group里。
Flask 2:在这个烧瓶里我放置了15个杂双环化合物,部分来自于Ilib Diverse的”Heterocycles,bicyclic”,部分从Chem-Space搜索(参见:KNIME教程|基于反应的组合库生成)下载。
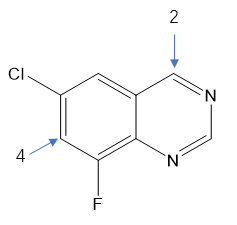
图6. 6-氯-8-氟喹唑啉的原子反应性设置
根据规划,双环片段设置两个连接点,分别与flask1与flask3的片段连接。双环片段是有方向性的,以6-氯-8-氟喹唑啉为例,设置了如图6所示的原子反应性:4位碳原子反应性为2,7位碳原子反应性为4,其它碳原子为0,以便确保该片段第一次反应时(即与flask1片段反应时)是以7位进行反应;并且设置7位原子“reset reactivity after first substitution”以确保第一次取代反应发生后该碳原子的反应性为0,防止发生二次取代。
如果想完全重现Bayer的结构生成过程,需要将原文附件的双环化合物全部导入,在本文中出于演示的目的,并没有使用文献提到的全部片段。
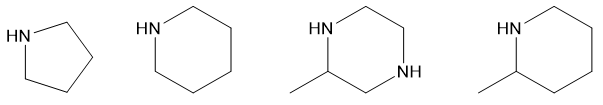
图7. 4个单环杂环化合物
Flask 3:使用了图7所示的单环杂环化合物,为了控制反应的方向性,也对N与C原子做了不同的反应性设置。
Flask 4:为共价结合弹头部分,在本算例中,仅用了丙烯醛作为弹头。当然,真实的研究中你可以使用共价弹头,比如Kim等人(2021)[3]在综述中提到的那些弹头。
步骤3. 过滤与结构生成
如图8所示,在生成结构之前可以设置过滤条件。Ilib Diverse提供了多种过滤器用于结构生成的预处理,在本算例中,我们用默认过滤器,即分子量100-700,同时将含反应性基团的化合物滤除。

图8. 过滤与结构生成
点击Preview预览生成的化合物以供调整生成参数;点击generate则生成终的结果,还可设置生成的化合物数(100),保存为smiles格式。浏览结果文件,发现化合物4(图9)与Mortier等人报道的化合物1非常相似(pos2的芳香环与pos4弹头部分存在差异),该化合物由几个已知抑制剂对应的片段按预先设置的规则生成。化合物1对KRASG12C与KRASwt的IC50分别为2.8μM与大于10μM。
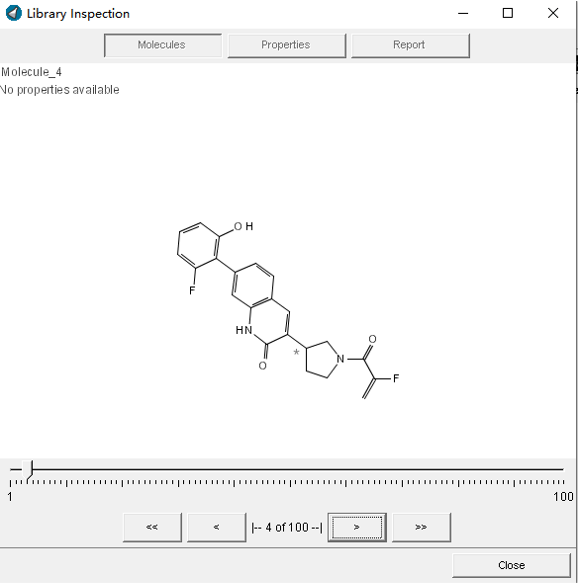
图9. 结果预览,上图为其中一次看到的4号化合物
这里,提供其中一次生成的100个化合物以供大家参考(每次生成的化合物可能会不一样),下载:KRAS-100.smi
3. 小结
本文用Ilib Diverse重现了拜尔公司Mortier等人的计算赋能工作流识别KRASG12C共价抑制剂中的组合库设计工作流。总的来说,Ilib Diverse使用非常简便,通过对试剂/片段的收集,以及自己对项目的理解,可以随时快速的枚举自己感兴趣的化合物库,最终实现加速项目进度的目的。
4. 接下来可以做什么
- 收集更多专利、科技期刊、会议报道的化合物片段,用于生成结构更多样的化合物库
- 用LigandScout、OEDocking、Flare等进行共价抑制剂虚拟筛选
- 用炼金术法FEP计算预测化合物的结合自由能
- 基于反应的组合库设计
- 利用深度学习(AI)重新生成更多的新化合物
目前,国内有很多公司通过基于深度学习方法进行训练、生成化合物库,再药物化学专家通过CADD手段进一步手工筛选,最后推荐出优选的化合物。如果用Ilib Diverse,不论是实验药物化学还是计算药物化学专家,也不论你是否懂AI结构生成,你仅需要在对科技文献、专利 或会议报道化合物进行收集、整理,就可以在需要的时候生成结构多样、高质量的组合库。Ilib Diverse是您低成本的药物专业助手。
Schulz等人(2018)报道了用LigandScout进行共价抑制剂的虚拟筛选,发现靶向肠病毒3ClPro Cys的共价抑制剂:Ligandscout案例 | 共价键结合药物的虚拟筛选
Nathan Kidley的算例演示了如何用Flare对接计算进行共价抑制剂设计:BTK共价抑制剂的设计
还介绍了如何用OEDocking等分子对接软件进行共价抑制剂的虚拟筛选:用常规分子对接进行共价抑制剂的虚拟筛选。
Flare支持绝对结合自由能计算与相对结合自由能计算,前者用Flare/waterswap实现,后者用Flare/FEP实现。
如果你对结构类型有偏好,可以预先构思好合成这类化合物的反应路线,根据该路线用实际可获得的试剂生成化合物库,具体的实例请参见基于反应的组合库生成。
你可以将上述生成的化合物、或经过药效团过滤、甚至Docking打分过后的化合物作为深度结构生成(比如深度迁移学习,增强学习)的训练集化合物,用获得的深度结构生成模型帮助你生成更多的新化合物以便用于下一步的虚拟筛选。
LigandScout作为成熟的药效团技术与成熟的深度学习从设设计平台(比如阿斯利康开源的REINVENT[4,5]等)组合,很容易实现深度增强学习的设计方案。比如,日本英德知的Yoshimori等人将LigandScout与REINVENT组合,成功地将药效团作为深度增强学习的打分函数(奖励函数)实现TIE2抑制剂与DDR1抑制剂的从头设计[6,7]。
5. 补充:基于药效团的虚拟筛选
5.1 基于结构的药效团模型识别
用Ligandscout基于结构的方法从PDB 6TAM识别药效团,结合基于结构的相互作用分析,确定如下图10的药效团模型。包含了如下几个必需的药效团特征:1)2个芳香中心(蓝色带箭头的环),图10左边的芳香环对应于组合库设计时规划的pos1部分,而右边那个对应于规划的pos2(图2)的一部分,其中与pos2对应的芳香环还与ARG68发生有利的cation-π相互作用;2)氢键供体(红色箭头)与受体(绿色箭头),对应于组合库设计时规划的pos2(图2)的一部分与残基GLY10发生相互作用;3)共价键相互作用特征(橙色球),与残基CYS12发生共价结合,对应于组合库设计规划的pos4部分(图2)。
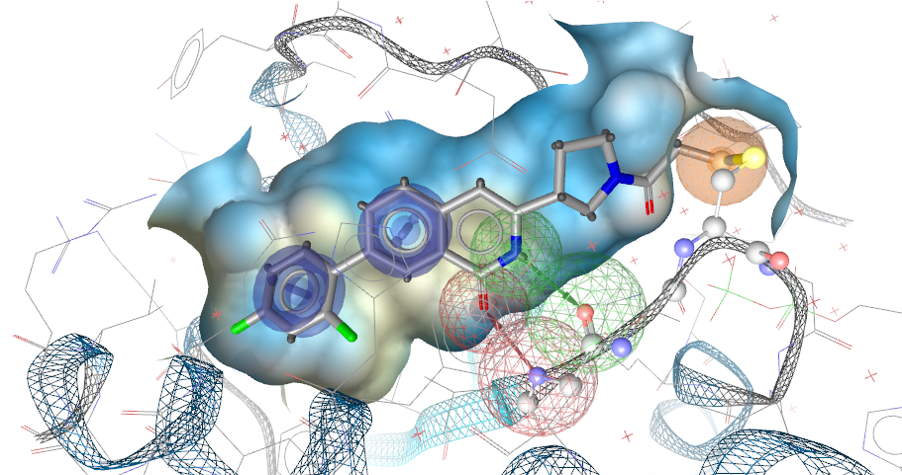
图10. 基于结构的药效团模型(从PDB 6TAM用Ligandscout生成)
一方面,出于演示目的,并没有对药效团模型进行严格的模型验证。另一方面,考虑到该药效团模型与组合库规划的设想一致,即使没有验证,也可以用于搜索满足规划要求的化合物。
应该要注意的是,Mortier等人(2020)[1]启动项目的时候,PDB 6TAM还未公开,是将ARS-1620对接到PDB 4LV6结合位点里进行分子动力学模拟,从轨迹获得的复合物结构再识别的药效团模型,这个药效团模型与图10的模型有非常大的差异,尤其是ARS-1620与残基GLY10之间没有PDB 6TAM所揭示的氢键相互作用特征。
5.2 化合物库的准备
出于演示的目的不需要生成非常多的化合物。因此,图2的pos2部分仅用了部分双环结构,大部分的双环权重被调低以便让更加接近于期待的目标化合物以更大地概率生成。同时,对图2的pos3部分也进行了权重调整,只让少部分环参与库的生成。这里仅仅输出了其中两个有较大差异设计策略生成的化合物,并保存为SMILES格式文件。
生成的化合物用OpenEye软件包依据《OpenEye | 化合物数据库的准备》描述的方式进行结构准备。其中重点步骤包括:1)去重复结果;2)进行互变异构体枚举(通过tautomers命令来实现);3)对未明确定义手性的手性原子进行立体化学枚举(通过Omega的flipper参数来实现);4)用omega生成一个3D低能构象。最后得到的3D结构保存为sdf格式以备给后用。
Ligandscout的database generation用于最后的构象搜索与数据库准备。具体而言,icon以best模式读入上一步omega获得的3D结构准备数据库于需要筛选。准备好的数据库分别保存为kras-strategy01.ldb与kras-strategy02.ldb。
5.3 虚拟筛选
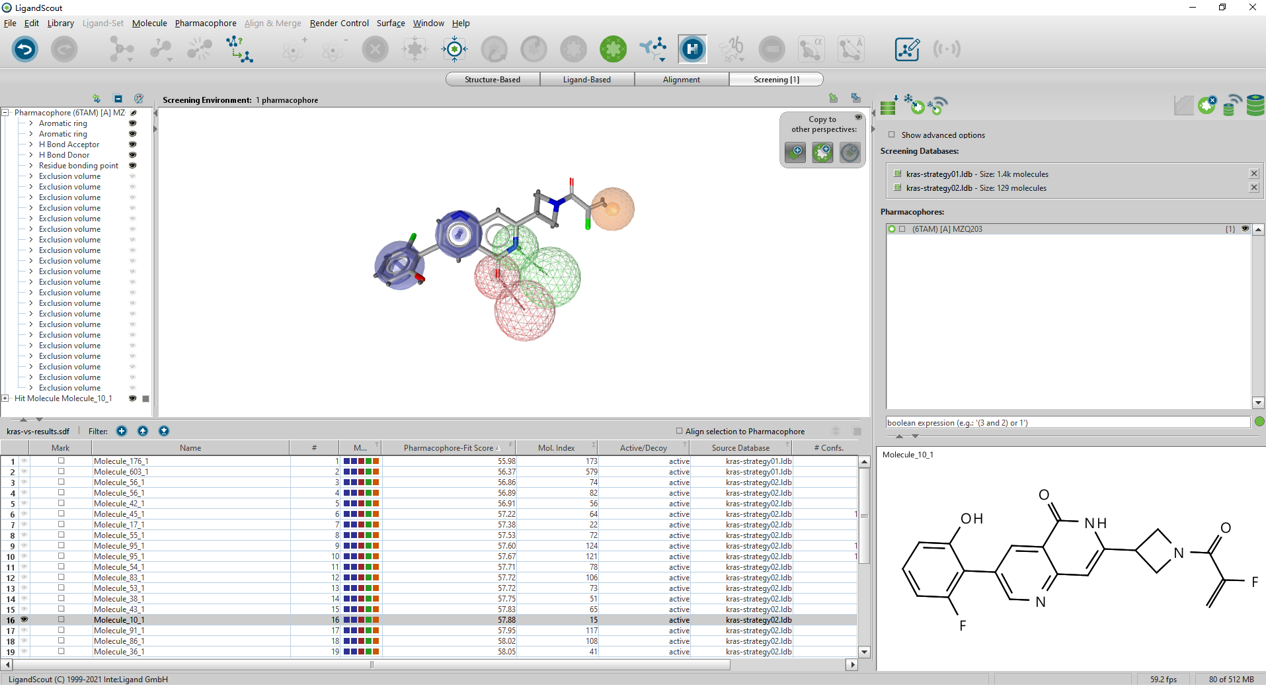
图11. 在Virtual Screen里读入药效团与两个数据库,进行虚拟筛选命中34个化合物
在ligandscout里将5.1与5.2准备好的药效团、数据库读入到Virtual screen模块里,采用默认参数进行虚拟筛选,结果命中34个化合物。两个典型的命中化合物结构与药效团匹配的结果如图12所示。
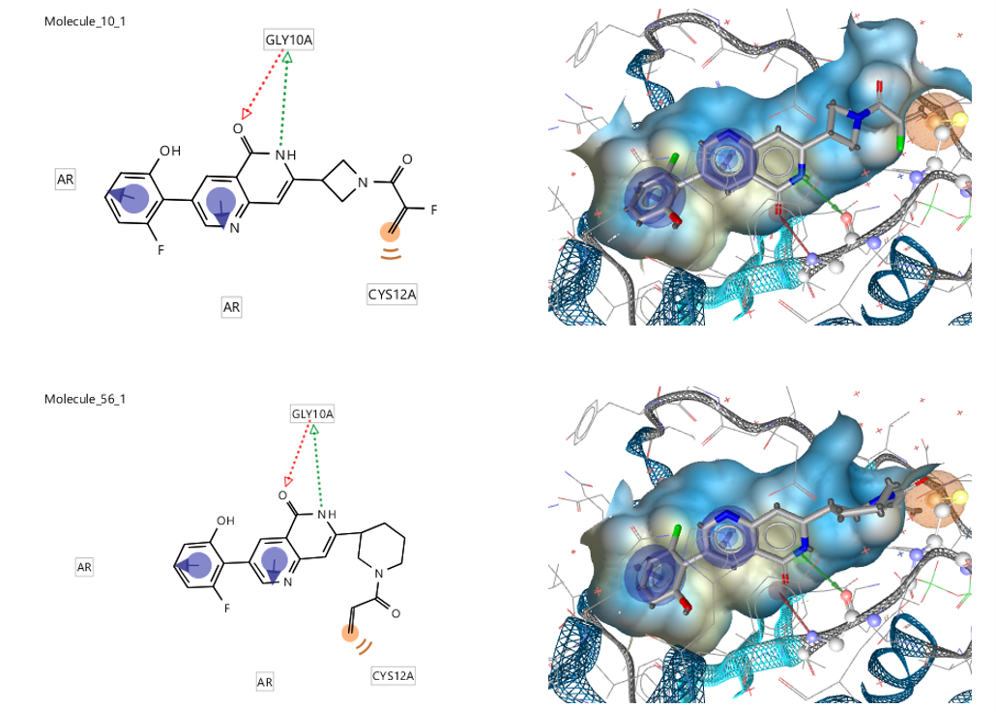
图12. 虚拟筛选命中的两个典型化合物结构及其与药效团的匹配
更有挑战的场景是如何主要利用PBD 4LV6的信息进行虚拟筛选,一个有潜力的方法是共价分子对接。共价对接虚拟筛选的成败受4LV6结合位点内残基(比如MET72)的柔性侧链影响。MET72是一个柔性的残基,会因配体原子的出现而改构象。在4LV6结合口袋中,MET72的侧构象并不适合于诸如6TAM共晶配体之类化合物的对接计算。因此事先将该残基构象进行变化以便容纳配体原子是共价对接虚拟筛选的关键。合适的MET72侧链构象预处理可以将PDB 6TAM共晶配体在4LV6结合位点里摆出正确的pose(cross-docking)。在用Flare/Covalent docking进行的一个测试中,6TAM的共晶配体被正确的cross-docking到4LV6的结合位点里(图13),其余34个药效团命中的化合物的对接结果也能重现之前药效团虚拟筛选的结合模式。
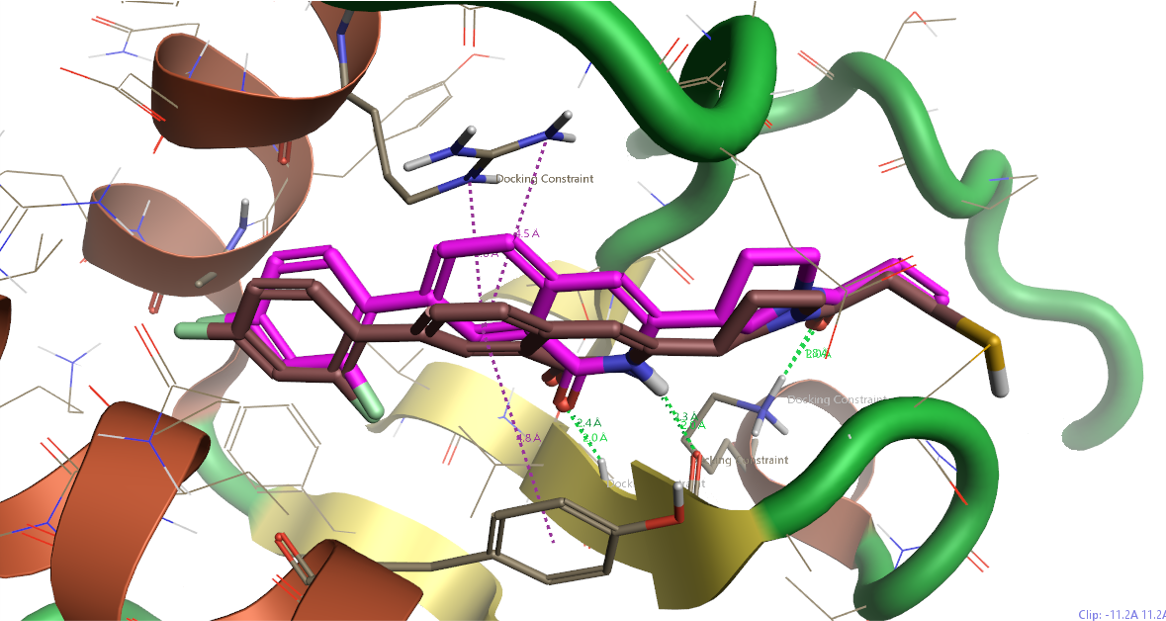
图13. 6TAM共晶配体(洋红色)及其cross-docking到4LV6结合位点的pose(棕色),计算与共晶的pose重合的相当好,发生偏移的部分主要是因为4LV6的ARG68侧链对配体挤压产生的。
5.4 相关附件下载
虚拟筛选用的药效团模型:6tam.pml
虚拟筛选用的化合物库:kras-vs-dbase.zip
虚拟筛选命中的化合物:6tam-vs-hits.sdf
6. 文献
- Mortier, J.; Friberg, A.; Badock, V.; Moosmayer, D.; Schroeder, J.; Steigemann, P.; Siegel, F.; Gradl, S.; Bauser, M.; Hillig, R. C.; et al. Computationally Empowered Workflow Identifies Novel Covalent Allosteric Binders for KRASG12C. ChemMedChem 2020, 15 (10), 827–832. https://doi.org/10.1002/cmdc.201900727.
- L. Li, J. Feng, T. Wu, P. Ren, Y. Liu, Y. Liu, Y. O. Long (Araxes Pharma LLC), WO2015054572, 2015.
- Kim, H.; Hwang, Y. S.; Kim, M.; Park, S. B. Recent Advances in the Development of Covalent Inhibitors. RSC Med. Chem. 2021. https://doi.org/10.1039/D1MD00068C.
- Olivecrona, M.; Blaschke, T.; Engkvist, O.; Chen, H. Molecular De-Novo Design through Deep Reinforcement Learning. J. Cheminform. 2017, 9 (1), 48. https://doi.org/10.1186/s13321-017-0235-x.
- Blaschke, T.; Arús-Pous, J.; Chen, H.; Margreitter, C.; Tyrchan, C.; Engkvist, O.; Papadopoulos, K.; Patronov, A. REINVENT 2.0: An AI Tool for De Novo Drug Design. J. Chem. Inf. Model. 2020, 60 (12), 5918–5922. https://doi.org/10.1021/acs.jcim.0c00915.
- Yoshimori, A.; et al. Strategies for Design of Molecular Structures with a Desired Pharmacophore Using Deep Reinforcement Learning. Chem. Pharm. Bull. 2020, 68 (3), 227–233.
- Yoshimori, A.; Asawa, Y.; Kawasaki, E.; Tasaka, T.; Matsuda, S.; Sekikawa, T.; Tanabe, S.; Neya, M.; Natsugari, H.; Kanai, C. Design and Synthesis of DDR1 Inhibitors with a Desired Pharmacophore Using Deep Generative Models. ChemMedChem 2021, 16 (6), 955–958.
7. 测试
如何你需要测试Ilib Diverse,请准备一张PPT,描述一下您的库生成规划或策略(如图2所示);再用ChemDraw画出你计划采用的片段,并保存为mol格式文件(注意不是MOL V3000格式),每个烧瓶(flask)一个文件夹。然后,我会帮你生成化合物库。


