
基于结构药物发现中的决策:对接结果的手工检查
摘要:分子对接是一种广泛应用于药物发现的计算方法。由于分子对接固有的不精确性,结合模式的手工检查是计算药物化学家在决策过程中至关重要的例行程序。然而,尽管它对药物化学研究项目十分重要,但对接姿势(pose)的手工检查的指南在文献中却鲜有讨论。在此,我们回顾了药物化学相关文献,以确定手工检查的一致原则,重点突出其中成功应用的案例,并且讨论它的局限性。在此,我们对学术界和制药行业的专家进行了一项调查,其中还包括一项区分自然姿势和错误姿势的挑战。我们收集了93项专家意见,为手工检查支撑的决策过程提供有价值的见解。这篇综述可启发经验丰富的计算药物化学家们的讨论,并指导新进入该领域的年轻科学家对他们的复合物进行分析预测。
原文:Fischer, A.; Smieško, M.; Sellner, M.; A. Lill, M. Decision Making in Structure-Based Drug Discovery: Visual Inspection of Docking Results. J. Med. Chem. 2021, 64 (5), 2489–2500. https://doi.org/10.1021/acs.jmedchem.0c02227.
编译:李凯丽,张丽君,柏川(中山大学)/2021-06-04
介绍
基于结构的大型化合物数据库的虚拟筛选以及配体结合模式评估等计算手段,已成为现代药物发现的重要组成部分。它们不仅可以提供可能与特定靶标结合的配体,而且还可以解释观察到的现象,如化合物的构效关系。因此,基于结构的计算方法可以用在药物发现和开发的各个阶段。虚拟筛选的普及源于其低成本高效益、识别新的化学结构的能力,以及对配体-蛋白质相互作用机制的理解。分子对接是基于结构的药物发现的主要方法之一。在分子对接中,可以确定有利的配体-蛋白质相互作用的构型,产生所谓的配体姿势(pose,结合模式)。它们与蛋白质的相互作用可被打分函数量化。为了实现分子对接的效率,会通过一个简单的打分函数来量化配体-蛋白质相互作用。但是这种简化的蛋白质-配体相互作用模型是该方法的一个很大的限制,尤其表现在对姿势排名的不准确,并且在预测绝对或相对结合自由能方面表现不佳。此外,配体结合模式的正确采样可能受限于诱导契合效应以及蛋白质的不同构象状态(蛋白质通常被视为刚性体),这些因素导致的不精确度超过打分函数自身所带来的不精确度。此外,有时分子内相互作用的模型是错误的,比如出现扭曲的酰胺基团、蛋白内配体之间的结构冲撞,或不现实的三维(3D)结构。而影响分子对接的另一个关键因素是输入结构的分子质量。常见的缺陷和注意事项包括,配体的质子化状态,互变异构体形式,结合位点的酸性和碱性残基的特殊质子化状态,以及谷氨酰胺、天冬酰胺和组氨酸残基的翻转。在虚拟筛选应用中,对接方法经常会产生大量假阳性结果,从而限制了其作为独立平台的适用性。很多方法也被用来改善分子对接的结果,例如,通过运行分子动力学(MD)模拟,然后进行MM/GBSA(molecular mechanics/generalized Born surface area)、炼金术结合自由能计算(alchemical binding free energy calculations)或现代机器学习方法等,但上述做法的效果都不佳。由专家检查对接结果,尤其是对接结果预测的结合模式,对于成功识别苗头化合物(hit)或先导化合物的优化尤为重要。最近有综述报道,在250篇已经发表的虚拟筛选工作中,约有50%在化合物的优先级排序中进行了手工检查。因此我们建议任何一个有适当经验的人都可以人为地评估对接姿势的好坏,而不仅仅是依靠打分得到结果。虽然如此,人们已经开发了多种计算方法和工具来辅助决策。他们采用了诸如相互作用指纹图谱、结构骨架对接或比较多个同系物获得的结合模式等技术来辅助决策过程。其中一些工具是最近才开发出来的,并且以Web服务器的形式提供。
如下所述,对接结果的手工检查已被用于几个成功的发现项目中。此外,手工检查也应用于对接实验方案的验证过程中,特别是通过重新对接(redocking)或交叉对接(cross docking)姿势预测进行评估,以完善均方根偏差(RMSD)和天然接触(native contacts)等评估标准。然而,大多数这些研究只是模糊地提出或者根本不提出所用的标准。最近的一篇综述关注了在药物化学中的决策方面,涉及对活性(potential),选择性,药代动力学,结构生物学和计算结果的大数据集的评估。在基于这些数据集的化合物优先级排序方面,作者报告了药物化学家之间存在相当大的分歧,可能是由于他们不同的经验、背景和培训。不同研究人员之间的这种差异也可能存在于对对接姿势的可视化评估中。因此,我们有必要全面回顾应用标准,以确定配体结合模型手工检查的一致准则。
在这里,我们回顾了药物和计算化学出版物,在这些出版物中手工检查被应用于补充虚拟筛选活动中的分子对接和姿势打分。我们报告了成功应用的案例及其局限性,以及其对化合优先级排序预测准确性的直接影响。此外,我们还对计算药物化学方面的专家们进行了一项调查,以确定他们在对接姿势的手工检查标准中达成的共识和存在的分歧。由此,本文旨在为本领域中进行对接结果的手工检查的实用性和最佳策略提供更广泛的讨论基础,并为该领域的新人或年轻研究人员提供指导。
手工检查标准
表1. 成功的手工检查用到的标准a
| 标准 | 靶标 |
|---|---|
| 结构新颖性,氢键网络,与特定残基的相互作用b,互补性,配体柔性,水相互作用,配体张力,疏水相互作用 | purine nucleoside phosphorylase |
| 结合模式一致性c,配体质子化,互变异构体、互补性、与特定残基的相互作用b | aromatase |
| 独特的相互作用d,与特定残基的相互作用b,配体柔性,与晶体结构的比较,结构多样性 | κ-opioid receptor |
| 与特定残基的相互作用b,疏水相互作用,配体张力 | RANKL receptor |
| 与特定残基的相互作用b,互补性,疏水相互作用,π-堆积,氢键网络, 水相互作用,配体刚力 | ecto-5′-nucleotidase |
| 扭曲的配体几何e,配体张力,溶剂暴露基团的性质,静电排斥,不满意的配体杂原子,疏水相互作用,π-堆积,阳离子-π相互作用 | enoyl-acyl-carrier protein reductase |
| 结构新颖性,氢键网络,溶解度 | enoyl-acyl-carrier protein |
| 不满意的配体杂原子,配体柔性,扭曲的配体几何,氢键网络,互补性 | EphB4 kinase |
| 互补性,与特定残基的相互作用b,氢键网络,疏水相互作用,π-堆积,结合模式一致性c | γ-glutamylcysteine synthase |
| 互补性,结合模式一致性c,不恰当的原子类型,亲水-疏水错配,溶剂暴露基团的性质 | Bcl-xL |
| 互补性,π-堆积,氢键网络,与特定残基的相互作用b | thymidine monophosphate kinase |
| 结构多样性,疏水相互作用,氢键网络,互补性 | β2-adrenergic receptor |
| 互补性,结构多样性,配体柔性 | aldose reductase |
| 与特定残基的相互作用b,π-堆积,疏水相互作用,,氢键网络 | N-myristoyltransferase |
a.发现生物活性化合物的研究以及在视觉检查中应用的标准。b.与特定氨基酸或辅因子的相互作用。c.多个对接配体结合模式的一致性。d.不存在于同源靶中的相互作用。e.扭曲的酰胺、酯和其它官能团。
在手工检查中,最常用的标准之一是化合物及其结合位点的空间互补性(如表1)。等温滴定量热实验揭示了掩埋的非极性表面与配体的结合自由能之间的关系。此外,在基于结构的理性先导化合物优化中,互补性的部分缺失可以用来指导配体的扩展(expansion)或修饰。而在一些研究中,手工检查还将形状互补性进一步区分为静电互补性和疏水互补性,加上蛋白和配体的物理化学性质,从而为复合物提供了更详细的的相互作用分析。
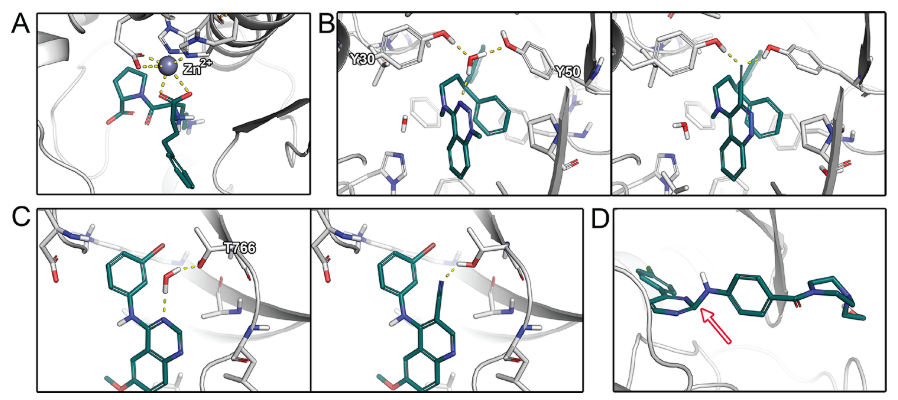
图1. 结合模式示例。 (A) 赖诺普利与血管紧张素转化酶的配体-金属相互作用 (PDB 1O86)。 (B) N-(3,3-Diphenylpropyl)-4-benz-1,2,3-triazine-amine (左) 和 3-cyano-N-(3,3-diphenylpropyl)-4-cinnolin-amine (右)在存在或不存在水分子的情况下与scytalone dehydratase结合。水分子被氰基置换导致结合自由能增加2.0 kcal/mol。该图在晶体结构(PDB 5STD)和水合位点预测研究后进行了调整。 (C) 4-Anilino-6,7-dialkoxyquinazoline (左)以及 4-anilino-6,7-dialkoxyquinoline-3-carbonitrile(右)与EGFR 激酶结合。水分子的置换导致不利的结合自由能。图 C 在晶体结构(PDB 1M17)和水合位点预测研究之后进行了调整。(D) 在晶体结构中的应变配体构象示例(PDB 3QAD),2-氨基嘧啶片段偏离平面(箭头所示)。此PBD结构因此进行了精修,具有更合理的构象(PDB 3RZF)。
绝大部分我们评估过的研究都报道了检查配体和蛋白质之间的氢键相互作用(如图1)。由于氢键作用相对较强,且氢键相互作用的空间几何要求清楚,它们很容易被手工检查,也是结合亲和力和选择性的重要因素。通常,手工检查通过对比共晶结构的配体或对比具有类似化学结构类型的配体,首先考察配体结合位点的氨基酸残基与配体之间产生的氢键作用网络。例如,在设计酶抑制剂时,与催化残基的特定相互作用对于有效抑制催化反应和抵抗潜在突变非常重要。一般来说,根据环境的不同,不同的氢键强度、稳定性以及最终对结合亲合力的影响会有很大的不同。疏水性蛋白质环境中的氢键通常比溶剂区域中的氢键更有利于结合的自由能,因为在两种环境中去溶剂化自由能及其静电相互作用的衰减存在显着差异。这个性质在有电荷参与的氢键,也就是盐桥中尤其明显。例如,由于带电荷的氨基酸残基的去溶剂化所耗能量高,结合位点中暴露于溶剂表面的盐桥对结合自由能的贡献通常可以忽略不计。与侧链原子形成的氢键相比,蛋白质骨架的氢键通常更强,通常较少受到侧链突变的影响。对于主链上的氢键来说,蛋白质的熵损失通常较小,因为侧链比主链自由度更大。在氢键形成时,配体-蛋白质复合物的刚性化常伴以配体和蛋白的自由度降低,从而导致熵减少。这个性质可以被利用,即改造刚性配体部分来产生多个协同氢键来减少熵-焓补偿的影响。因此,氢键的位置对于实现有效的配体结合是至关重要的,在手工检查分析时应该仔细考虑蛋白质环境对这种相互作用的影响。除了已形成的氢键外,还需要评估所有没有对应相互作用对象而未被满足的氢键供体与受体基团(特别是那些被埋在结合位点里的那些),因为它们通常会由于未被补偿的去溶剂化自由能损失而导致结合自由能的减少。在金属蛋白中,与金属辅因子独特的静电相互作用对配体的结合亲和力具有重要意义(图1A)。有趣的是,在本综述涉及的研究中,没有将弱氢键、卤素键、正交偶极子相互作用或酰胺- π相互作用作为手工检查的标准(其他细节见调查结果)。
溶剂化效应是蛋白质-配体结合热力学过程的主要贡献者。在配体结合过程中,水分子在溶剂化腔中被置换出来。虽然由于熵的增加(有时是焓)而使水分子的置换有利,但有些分子可能与蛋白质形成焓有利的相互作用,包括直接与蛋白的相互作用或帮助形成配体-蛋白相互作用,从而有利于结合自由能。因此,在手工检查中应该考虑溶剂化效应,如水介导的相互作用(图1B,C)。但是,对水的多层次行为的正确评估对于专家也不是一件容易的事。此外,未结合配体的蛋白的结合位点中的溶剂化情况通常不在手工检查之列。可以使用其他软件来估算水分子置换的焓和熵的贡献,例如,基于MD模拟或派生的神经网络,可以增加对接和打分的准确性。另一个相关的评价标准是暴露在溶剂中的配体原子的性质,突出到周围溶剂的疏水性基团通常被认为是不利的,因为它们在其它姿势中将有机会与蛋白相互作用。
而在大多数配体-蛋白复合物的形成过程中,两者都必须调整它们的构象以优化它们的互补性。据估计,25%的蛋白质在配体结合过程中经历了一定程度的诱导契合。随后在蛋白质数据库中对188种蛋白质进行了研究,这些蛋白都具有结合配体的结构和未结合配体的结构,研究显示其中90%的蛋白质在结合后会发生旋转异构体(构象异构)变化。配体或蛋白质变形时的这种能量消耗称为张力,而打分函数通常不能很好评估张力的能量变化。此外,由于在大规模虚拟筛选所使用的分子对接计算中蛋白质通常是刚性的,因此两个结合配偶体之间无法相互适应。两种缺点都可能导致对接产生的配体内部几何形状不切实际(图1D)或接触过度。例如,观察到扭曲的酰胺或酯基团,导致在手工检查时拒绝了这些的结合模式。因此,在全面的手工检查中应考虑配体和蛋白质的张力。因为处理蛋白张力的前提是对蛋白结构进行柔性化处理,所以应该使用柔性的对接和基于MD的后处理来考虑蛋白质和配体的构象互配。但是,对于大量的配体-蛋白质复合物而言,这在计算上仍然过于昂贵,例如虚拟筛选就存在这个问题。但在先导物优化中,由于化合物的数量较少,这类方法的使用往往变得容易。
蛋白-配体复合物的现有知识可能有助于确定结合姿势。结晶结构中已知配体与靶蛋白的相互作用的知识,可以直接用于检测新的配体-蛋白相互作用。结果表明,与天然配体的对接的训练提高了结合姿势预测的准确性。此外,由于同源靶标通常具有保守的配体和结合模式,因此在决策过程中可以考虑具有相似共结晶配体的相关蛋白的晶体结构。
手工检查在计算机辅助药物设计中的应用案例
如上所述,手工检查已经应用于大量的药物发现项目中。在大多数情况下,这个检测是在进行实验测试之前选出最优化合物的最后一步。在表1中,我们总结了几项研究,这些研究提供了用于手工检查标准的详细说明,利用这些标准发现了高活性的配体化合物。正如前面提到的,手工检查中最常用的评估对象是配体和各自结合位点之间的互补性,以及配体与蛋白质之间的相互作用,如疏水作用和氢键。此外,也经常考虑与特定结合位点残基或辅助因子的相互作用。考察这些特殊的相互作用大多源自共结晶中配体和蛋白的相互作用,或者是源自与不同配体与同一蛋白产生的相同相互作用。而扭曲的配体构象也经常被认为是对接姿势的排除标准,因为它们代表不现实的配体张力,例如酰胺键、酯或不利的顺反异构。
相反,在相关的研究中很少提及诸如与保守的或结晶结构中的水分子的相互作用,以及与配体选择性相关的相互作用等特征。而化学多样性,新颖性和得分最高的配体的商品可得性不是手工检查的直接部分,而是对接结果进行后处理的标准。
在药物设计的筛选评价环节,尽管手工检查是其中的重要组成部分并被提及,但少见对手工检查的细节描述。应该详细描述在药物研发决策过程中的手工检查标准细节,因为这样可以保证结果的可重复性,这是发表计算机辅助药物设计(CADD)研究的常见要求。
接下来,我们重点介绍一些最近的研究,这些研究展示了手工检查在阐明结合模式中的应用。例如,一项大规模的虚拟筛选研究发现了可与AmpC β-内酰胺酶和D4多巴胺受体结合的配体,该研究使用了一个由1亿7千万种化合物组成的计算库。作者不仅使用手工检查发现了具有新化学类型的高效配体,而且还比较了使用或不使用手工检查筛选出的配体。有趣的是,后者的分析表明,基于手工检查后选择得到的配体通常表现出更好的结合亲和力,由此证实了这种方法的实用性。
表2. 人工检查的直接评估

药物设计数据资源(D3R)定期邀请计算化学和CADD领域的专家参与挑战,即以盲法预测配体结合姿势,对配体进行排名并预测相对结合自由能。虽然在第二次挑战中表现最佳的结果没有进行手工检查,但在其余挑战中表现最佳的结果通常依赖于人为干预(表2)。同样,在先前的SAMPL1挑战赛中表现最好的参赛作品也使用了手工检查。参加了SAMPL4挑战的参与者发表的一项研究提供了有关决策过程的详细信息,并回顾性评估了他们在人为干预和不干预情况下的筛选结果。发现当手工检查与打分相结合时,可以达到最佳的命中率。在D3R之前, Community Structure−Activity Resource ( CSAR )也有过类似的挑战,但只是简单地提到了手工检查,没有与未经人为干预的结果进行比较。
人为干预的局限性
尽管上述研究强调了人为干预在基于结构的药物发现研究中的优势,但这种做法也有局限性。最明显的是,进行手工检查的个人或团队的专业水平和直觉强烈影响了其结果,我们对手工检查的评估也证实了这个问题。另一个局限是在有限时间内手工检查的结合模型的数量。虽然一些研究报告检测了小于100个结合模型或复合物,但在大多数研究中检测了100到300个复合物。还有一些研究报告了对不到1500个化合物的测试,这很可能是手工检查应用的上限,因为如此大量复合物的手工检查会变得非常耗费时间,更困难的是在长时间跨度下保持评估的一致性。近期报道单个研究项目筛选的化合物库可包含多达数十亿种化合物,使得单个项目中复合物库的规模不断扩增,手工检查可能将难以实施,或者必须用其他技术来缩小范围。然而,现有评估的研究没有注明手工检查的各项任务完成的时间表。
此外,研究所使用的对接方法存在固有的局限性,单凭手工检查并不能抵消。例如,蛋白质的柔性在配体结合中起着重要的作用是一个共识。例如,众所周知,蛋白质的柔性在配体结合中起着重要的作用。尽管柔性对接引擎取得了进展,但大多数大规模的对接实验仍然没有将蛋白质的柔性和结构适应性考虑在内。因此,手工检查也是基于形成的晶体结构的复合物的静态视角。除此之外,构型导致的熵减变化,或去溶剂化导致的自由能的损失很难用手工检查来评估。然而,经典的打分函数主要是使用与配体-蛋白质结合相关的相互作用的简单数学式来评估焓贡献。熵效应,在打分函数中表现得很差,主要局限于配体去溶剂化(例如疏水接触)和配体构象熵(例如可旋转键的数量)的简单评估。这种对结合过程热力学的不当处理在对接过程中产生了一个重要的局限性,例如,补偿熵贡献可以显著降低结合亲和力。由于单靠手工检查无法正确地研究这些影响,因此需要结合配体-蛋白质结合的更动态表征,例如分子动力学(MD)模拟。MD模拟可以从本质上描述蛋白质和配体的灵活性,并且基于包含熵效应的轨迹后处理进行评估,因此与简单的打分函数相比,可以提供更精确的结合自由能的评估。MD模拟也可以用来提供配体-蛋白质复合物稳定性随时间变化的示意。研究表明,与MM/GBSA计算或推导相结合的MD模拟可以提高对接实验中姿势亲和力的预测,虽然其结果具有高度的案例特异性。目前,自由能微扰(FEP)被认为是计算结合自由能最精确的方法,其精度约为1千卡/摩尔。尽管最近在溶剂化效应的改进处理及其在神经网络后处理方法中的集成方面取得了进展,但在对10个对接项目的大规模评估中发现,标准的对接程序产生的最好得分姿势结果,通常也只能在2A的RMSD阈值范围内重现60%的天然结合姿势。这就强调了有必要进行手工检查以排除非天然姿势。
对计算药物化学家的问卷调查
在介绍中,我们回顾了药物化学家们在复合物优先级判定方面存在重大分歧,但是考虑到这个问题的高度复杂性,产生这些分歧也并不奇怪。为了更详细地展示目前手工检测的现状,我们对该领域学术界和制药行业的专家进行了调查。我们总共收集了93个专家意见。调查包括三个部分:(i)关于参与者的信息,如专业和经验,(ii)关于手工检测、所使用的方案和决定标准的普遍问题,以及(iii)识别三种配体−蛋白复合物的天然结合模式的挑战。我们通过电子邮件或领英单独联系了调查的候选参与者。所有的参与者最近都在同行评议的期刊上发表了基于结构的药物发现领域的文章。调查表是由谷歌提供的指定平台制成的。此外,我们还鼓励参与者将该调查分享给他们有经验的同事。

图2. 问卷调查结果. (A) 调查参与者的科学职位分布。 (B) 调查参与者在计算机辅助分子设计方面的从业年数分布。 (C) 所有参与者为验证所使用的对接方法而采取的流程。(D) 所有参与者用于对接结果后处理的方法。(E) 所有参与者、制药行业参与者和其它参与者对各种标准重要性打分。 (F) 与总体结果相比,不从从业年数的(所有)参与者对不同标准的重要性打分。
大多数反馈者来自学术界(71,76.3%),其次是20名(21.5%)来自制药行业的专家,而只有两名来自非营利组织或政府机构。最大部分的参与者是计算化学家,其次是药物化学家和化学信息学家(图2A)。报道中超过一半的参与者在计算机辅助分子设计领域有超过10年的经验(图2B)。这相当于超过600年的经验。从1分到5分的范围,参与者对药物设计项目中可视化检测的重要性打分给到了4分(29.0%)或者5分(64.5%),这强调了这类程序的普及程度。
在对接和可视化检测之前,通常建议验证所使用的对接实验方法,以追溯评估姿势预测的准确性,以及区分活性化合物与参比无活性化合物的能力。尽管大多数参与者(80.6%)声称,他们将共结晶配体重新对接到x射线蛋白结构中,但其中大约只有一半的参与者将多种配体与所选的蛋白质结构交叉对接,或使用无活性的参比化合物来确定富集活性化合物的标准(图2C)。令人惊讶的是,有11名参与者(在学术界和工业界之间平均分布)没有执行上述任何验证步骤。
大多数参与者并不把对接作为唯一的计算方法。通常,他们会对对接结果进行后处理,例如,通过使用MD模拟,来应对标准对接方案中对蛋白质柔性的局限性,并评估所获得姿态随时间变化的稳定性。关于手工检查,大多数参与者不仅自己进行个人评估,还会在团队中和同事讨论结果进一步检查。大约三分之一的参与者只由自己进行手工检查,与此同时,有10名参与者(10.8%)只与同事一起评估结合模式。有趣的是,当我们将参与者分为学术界和企业部门时,行业中只有12.7%的参与者和同事一起进行结合模式的评估,这比学术界(4.6%)占的比例多。在同事委员会之间的个人和集体意见的整合,利用了单一研究人员的经验和一个团队的卓越表现,从而提高了检查的质量。
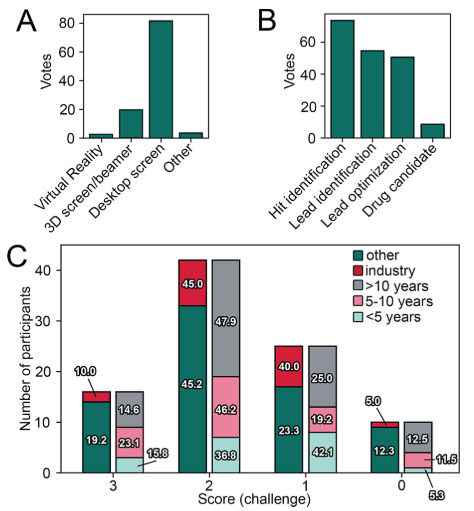
图3. (A) 参与者用于可视化的硬件。 (B) 在研发现管线中的参与者应用了视觉检查的任务。 (C) 根据隶属关系和经验划分的参与者给出正确答案(分数)的数量。 百分比表示被归类为一组的参与者(例如,来自行业的参与者)达到一定分数的总人数的比例。
尽管近年来在CADD环境中开发探索虚拟现实(VR)的步伐不断加快,为人类与3D数据交互提供了新的可能性,但迄今为止专家对VR的应用仍然有很大的局限性(图3A)。在我们的调查中,超过88%的参与者是在普通台式电脑屏幕上进行可视化检查。21.5%的参与者在VR和常规屏幕界面上使用了3D屏幕或3D激光仪。在药物发现过程中的不同时间节点上,从发现苗头化合物到最终候选药物,手工检查的受欢迎程度不断降低(图3)。
在调查中,我们评价了用于评估结合姿势的不同标准的重要性(图2E、F)。与之前讨论的研究一致,形状互补性在最相关的标准中十分重要,这一点不依赖于评估者的经验或其所属工作单位性质,这可能是因为它第一眼就能被简单推导出来。共结晶配体的预测结合姿势与实验确定的晶体结构进行类比也是同样重要的。有趣的是,制药行业的科学家优先考虑应用结晶结构数据,可能是因为他们更容易拿到好的数据。配体−蛋白与特异性结合位点残基的相互作用在我们的调查和所涉及的出版物中都被高度重视。有趣的是,相较于学术界,制药行业的科学家更重视配体的类型,配体−蛋白与结合位点相互作用的位置,以及不适宜的极性配体或蛋白质杂原子。最有经验和在制药行业工作的参与者都十分重视不适宜的蛋白质-氢键供体或受体。然而,我们的问卷并没有区分出在结合位点的不恰当的产生氢键的基团,例如处于结合口袋深处的未暴露基团,它的去溶化效应必须通过有利的相互作用来弥补,而对于那些暴露于溶剂中的易于溶剂相互作用的极性基团则不用考虑这个问题。经验丰富的参与者更重视溶剂化效应,如配体−蛋白结合过程中水分子的置换。最值得注意的是,打分函数产生的对接分数最不受重视,特别是对制药行业的专家来说。看来,已知的对接算法打分和排序的不可靠性使其在专家中不大受欢迎。
所有关于不同参与者组之间某些标准偏好差异的陈述都基于p=0.05水平的双侧t检验,显示出统计显著性。测试结果用scipy python工具包处理。参与CADD项目的科学家,特别是刚进入该领域的新手,可能会从我们的调查统计结果中获益,因为这为他们提供了指导,并且他们可以将自己的程序和标准与该领域的其他科学家进行比较。
结合模式预测挑战
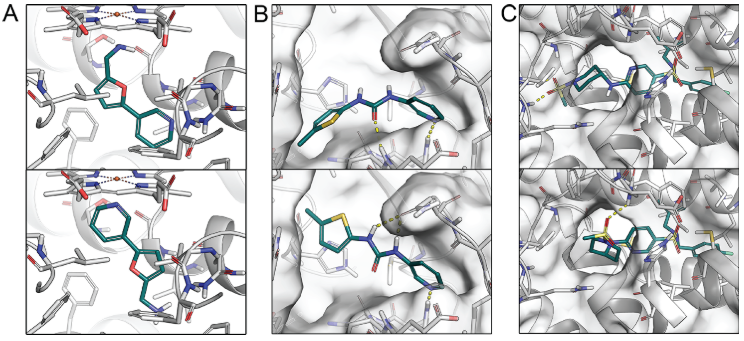
图4. 在调查中包括的结构,用于区分自然姿势和人工制品。 在调查中,这些结构以动画GIF图像的形式呈现,显示了所有相关的交互作用。 (A)案例1:5-pyridin-3-ylfuran-2-yl)methanamine与CYP2A6(PDB 2FDW)的 自然结合模式(上)与手工摆的结合模式(下)。 (b)案例2:1-(5-methylthiophen-2-yl)-3- pyridin-3-ylurea与SARS-CoV-2的主蛋白酶(PDB 5RH0)的自然结合模式(上)以及对接计算的结合模式(下)。 (c)案例3:N-[(4-fluorophenyl)methyl]-N-(2-methylpropyl)-6-[(1-methylsulfonylpiperidin-4- yl)amino]pyridine-3-sulfonamide 与RAR相关孤儿受体 γ (PDB 4WLB)的自然结合模式(上)以及对接计算的结合模式(下)
如上所述,我们的调查包括三个挑战,重点是识别天然结合模式,其中“天然”指的是共晶结构中配体的姿势。第一个例子是(5-吡啶-3-基呋喃-2-基)-甲胺与细胞色素P450 2A6(CYP2A6)结合的复合物,其末端胺基与血红素铁配位(表3)。由于静电作用,特别是与金属离子的相互作用,与配体的非结合态相比,结合态的pKa值明显降低,导致胺在配合物中不带电荷。此外,正如蛋白质数据库所注释的那样,由于结晶环境的pH值为8.5,胺的质子化状态可能已经转变为中性状态。为了获得具有可行的非天然姿势的相互作用曲线,我们手动反转了吡啶氮与金属配位有关的配体的方向(图4A)。出人意料的是,略超一半的参与者将配体的非天然方向视为正确的姿势。在调查中,我们提供了选择这三个示例的理由。不出所料,当参与者选择非天然姿势的配合物作为答案时,通常会提到胺的非常规质子化状态。此外,杂环含氮环部分(例如咪唑和吡啶)通常被视为CYPs中血红素铁的配位伙伴。含有胺的化合物与血红素铁配位的结合模式显示了配体的酸碱性这一特性(此处为碱性,且脂肪族的胺要比芳香环上的氮碱性强)如何影响与金属离子的相互作用,从而影响化合物的结合方式。
表3. 挑战结果与基于机器的排序
| 案例1 | 案例2 | 案例3 | |
|---|---|---|---|
| Correcta | 46.2% | 63.4% | 59.1% |
| wronga | 53.8% | 36.6% | 40.9% |
| Glide dockingb | correct | wrong | correct |
| smina dockingb | correct | wrong | correct |
| MM/GBSAb | correct | correct | inconclusivec |
a.关于各个挑战案例的调查结果。b.如果基于使用两个不同对接程序和MM/GBSA计算的结合能计算的排名确实对正确结合模式进行了优先性排序。c.自然的和不正确的结合模式在计算的能量上没有统计学意义上的显著差异。
因此,本案例代表了一种情况,就是大多数科学家的直觉和经验导致它们远离了正确答案。所以事实是,经验不足的科学家(只有不到五年的经验)比经验丰富的科学家更能选择到正确的结合姿势。选择天然结合模式挑战的参与者主要考虑配体-蛋白质相互作用的改善,包括吡啶环的氢键,以及质子化胺在口袋的疏水区域的不利位置。与其余的例子相比,在这种情况下,传统的打分函数和MM/GBSA计算优于人为排序。对于这些打分程序,配体的质子化如图4所示,天然结合姿势的末端胺呈中性,而在非天然复合物中末端胺则被质子化。以MM / GBSA方法的各个组成部分之间来看,天然结合姿势和非天然结合姿势之间最大差异在于静电和亲脂性作用。
第二个例子包括两种相似的1-(5-甲基硫酚-2-y基)-3-吡啶-3-叶脲与严重急性呼吸综合征冠状病毒2(SARS-CoV-2)的主要蛋白酶结合,其中一种是使用Glide SP对接方案(图4B)获得的。两种结合姿势形成两个氢键:天然姿势与蛋白质主链相互作用,而不是非天然姿势中的柔性侧链。在这种情况下,63.4%的参与者正确地识别出了天然姿势,是这三个案例研究中比例最高的。相比之下,这两个测试的对接程序都优先考虑了非天然结合模式,这支持了之前的假设,即专业知识优于传统打分函数。优先考虑天然姿势的参与者表示,形状互补以及与蛋白质主链间形成的更稳定的氢键作用是他们选择的决定性因素。相反,选择非天然姿势的参与者大多是基于配体的内部变化,并且通常是指氨基吡啶部分的构象。这一举例进一步强调了MM/GBSA计算的好处,因为它正确选择了天然姿势而不是所使用的对接方法。这一优势源于使用随时间变化的MD轨迹,并结合对溶剂化自由能更精确的处理。
第三个挑战包括一个反向激动剂与RAR相关的孤儿受体γ(RORγ)芳香族部分的两种相似的结合模式。然而,配体的磺酰胺部分以天然结合模式嵌在受体核心中,而在我们选择的非天然对接姿势中部分配体结构暴露于溶剂(图4C)。由于更好的疏水互补性和更有利的氢键连接网络,天然姿势更有利。有趣的是,尽管大多数参与者优先考虑了天然姿势,但仍然有超过40%的人优先考虑了非天然姿势。正如预期的那样,选择正确姿势通常是更大的氢键网络,以及配体与两个氢键更高的嵌合程度为理由。非天然姿势与不利的构象应变有关,因此在选择正确的姿势时不认为其是天然结合模式。然而,由于溶剂暴露的极性磺胺基团,非天然姿势被认为更有利。虽然两个对接程序都正确地预测了天然姿势,但MM/GBSA预测两种姿势具有相似的结合自由能。后者表明可能有多种有利的结合模式,但电子密度显示共晶结构中的配体原子的位置非常确定。基于20nsMD模拟的最后四分之一125帧的量化的MM/GBSA结合自由能的统计学显著性,在p=0.05水平有意义,该数据由scipy python计算包处理获得。虽然案例1和案例3的结果具有统计学意义,但案例2的平均值仅在p=0.07水平上有意义。
结论
由于分子对接和姿势打分的固有缺陷,在大量药物发现项目中需要对预测的蛋白质-配体复合物进行了手工检查。但它对实际决策过程的帮助也是有限的。我们回顾了可用的药物化学文献,来推导出应用标准,并总结了通过手工检查对接结合模式来直接评估预测准确性好坏。这些比较研究表明,在大多数情况下,手工检查能够提高预测性能,只有一项研究显示没有改善。最常用的手工检查标准包括形状互补性、氢键和疏水性接触。此外,我们还讨论了手工检查的局限性,包括可以准确评估的姿势数量、不同的经验水平以及基础方法的局限性,例如蛋白质的灵活性或熵的影响。为了更深入地了解该领域专家的决策能力,我们进行了一项调查,其中包括在三个案例研究中区分天然结合姿势和非天然结合姿势。在其它案例中,用未带电的伯胺代替抑制剂的杂环氮与CYP2A6中的血红素铁相互作用,以往使用CYP抑制剂的经验对天然姿势的检测产生了负面影响,这表明人类也可能存在偏差。大多数参与者表示,个人检查之后,与团队中的同事进行讨论,很可能会解决这种偏差并改善结果。与文献综述类似,被调查的科学家详细阐述了形状互补性,类似于在同结晶配合物中观察到的结合模式,且特异性的配体−蛋白相互作用是最有价值的标准。活跃在工业界中的科学家认为配体张力,不适宜的配体,以及蛋白质杂原子数量这些标准具有更高价值。众所周知,对接分数经常不准确,在可用标准中最不重要。这表明,走向可靠打分函数的旅程至今还没有结束,因为现今的打分函数通常与现有共晶数据和计算药物化学家的丰富经验是不一致的。总之,许多因素必须在手工检查时加以考虑,才能支持专家和该领域新科学家的决策。在调查结果的基础上,我们希望激励在同行评议期刊上评审对接研究的科学家将对接方案的验证以及包括手工检查和其他后处理方法作为常规要求。


