
摘要:如果采取适当的措施,对接预测的结合模式和亲和力是可以精确计算的。在柔性配体结合时,因构象自由度限制导致的结合能熵惩罚在计算上是困难的,但对于获得蛋白靶标与配体结合亲和力的可靠排序是非常重要的。
原文:David A. Winkler. Ligand Entropy Is Hard but Should Not Be Ignored. J. Chem. Inf. Model. 2020, 60(10):4421–4423. https://pubs.acs.org/doi/10.1021/acs.jcim.0c01146
编译:Irma/2024-03-28. 广州市墨灵格信息科技有限公司
药物发现中的高通量筛选、对接和打分
分子对接是一种广泛使用的方法,通过针对一个或多个蛋白靶标对大量候选物质进行筛选,以发现新药或重新利用现有药物。大多数虚拟筛选研究使用具有预定义打分算法的计算对接代码来对化合物进行排序。少数研究采用分子动力学模拟来模拟配体与靶标之间的相互作用,以获得更真实的对接模式和结合能。然而,很少有人进行广泛的计算研究,对构型空间广泛采样,以计算配体结合的吉布斯自由能的绝对值。
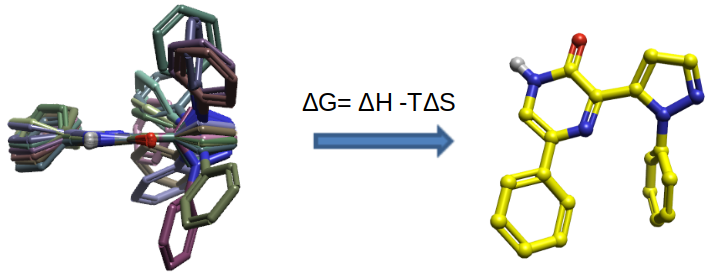
配体结合的吉布斯自由能ΔGbind定义为:
$$\Delta G_{bind} = \Delta H_{bind} – T\Delta S_{bind} = RTlnK_d = RTln{{[L][P]} \over {[LP]}} \cdots (1)$$
这里的ΔHbind是结合焓变,ΔSbind是结合熵变,Kd是解离常数,[L]、[P]和[LP]分别是配体浓度、蛋白浓度和复合物浓度(由实验确定)。
溶剂和配体柔性的熵贡献计算非常复杂,因此方程1中的TΔSbind项通常被忽略,或通过基于配体中可旋转键的数量或分子量的对数等相对简单的方法来近似。无法正确计算结合相互作用中的熵,是打分方法性能相对较差的原因之一,即使对接力场可以生成良好的结合模式。现在,由于大多数计算研究的目的是针对一个或多个靶标蛋白筛选多达数十亿的化合物,准确计算结合能所需的时间和资源通常是难以承受的1。
精确预测结合自由能难度大,且耗费大量资源,有时还忽视熵的贡献
准确计算结合自由能仍然是生物分子模拟中的一个挑战性问题2。最常用的方法包括基于热力学循环的炼金术法(自由能微扰,FEP);遵循物理路径的拉伸方法,使用一个力将配体从蛋白质中拉开;终点方法,如MM/PBSA与MM/GBSA;以及利用约束势能和偏置能对多个结合事件采样的元动力学(metadynamics)方法。最近,Aldeghi等人3报告了对一组结构多样BRD4抑制剂的绝对自由能的准确计算,该方法基于热力学循环。他们用X-Ray结构计算取得了平均绝对误差为0.6 kcal/mol,使用对接结构取得的平均绝对误差为1.0kcal/mol。然而,这种方法对计算要求太高,无法对大量候选药物进行筛选。绝对结合自由能评估需要对靶标蛋白和配体的构象空间进行广泛采样,这是一项对计算要求极高的任务。
由于使用绝对方法计算准确的ΔGbind的复杂性和资源需求,大多数计算筛选依赖于现有对接和打分算法,或对打分靠前的苗头化合物用PB或GB方法从MD模拟轨迹中计算结合能。一些广泛使用的对接和打分算法完全忽略了配体的熵,而其它一些算法则尝试通过近似方法来考虑这些熵惩罚项。
因此,配体构象熵的贡献通常被忽略,或者以非常近似的方式处理,这导致一些对接算法高估了柔性配体的结合能。这导致了柔性配体的排名高于其应有的位置,因为其与蛋白结合时构象自由度损失而导致的熵损失没有被考虑在内。
对打分靠前的对接苗头化合物的结合进行分子动力学模拟(MDS)可以改善性能
单独使用对接技术对于筛选大量化合物以识别出一小批有前景的候选物是有用的,但使用MD方法对这些苗头化合物进行后续模拟是明智的。在Guterres和Im近期的一项研究中4,使用高通量MDS显著改进了蛋白质-配体的对接结果。56个蛋白靶点(来自七种不同的蛋白质类别)和560个配体在ROC AUC的性能表现提升了22%,仅使用AutoDock Vina的ROC AUC为0.68,经MDS优化后的ROC AUC提高到0.83。然而,MDS比计算对接慢了几个数量级。Guterres和Im提出,这种改进是由于考虑了结合过程中的熵效应以及水分子的作用。
PB和GB方法在计算结合能时并不能可靠地考虑配体的构象熵
配体熵通常在分子动力学模拟中也没有得到很好的处理,这些模拟旨在从对接实验中提供对配体结合模式和蛋白质结合位点的结合自由能改善的预测。与MM/PBSA和MM/GBSA法结合使用的MD,经常被用来估算蛋白靶标与小分子配体的结合自由能。在准确性和计算工作量方面,上述方法介于经验对接打分和严格的炼金术微扰方法之间。虽然在考虑溶剂的熵贡献方面做得还算合理,但由于正则模分析的高计算成本,它们忽略或近似了配体的构象熵变化5,6。
显然,非常柔性的配体会受到熵惩罚,能显著影响它们的结合亲和力。因此,配体通常被刚性化以减少这种熵惩罚7。配体中可旋转键的数量是一个有用的参数,可以用来量化配体构象熵对ΔGbind的贡献。实际上,一些对接和打分方法使用每个可旋转键0.8−1.4 kcal/mol的经验值,来计算构象熵(ΔSbind)对结合能的贡献9。
看似矛盾的是,配体刚性化以减少柔性经常会使结合熵补偿性降低,而Gibbs结合自由能几乎没有变化。这种现象被称为熵-焓补偿9。从熵主导结合的配体到焓主导结合的配体,熵和焓之间的相互作用可以有很大的跨度。这在潜在药物的排序中造成了实质性差异,与更刚性的药物相比,大的、柔性的化合物在优先级排序中的位置下降了。
如何在高通量计算筛选中更好地考虑配体的构象熵?
对接和打分
对接程序中的打分函数分为三类:基于力场的、经验的和基于知识的。一些基于知识的打分方法,如ITScore,包括构象熵10。在拟合打分参数时,像LUDI和X-Score这样的打分函数在ΔG0回归常数中,隐含考虑了由于旋转和平移自由度限制而导致的熵损失。Surflex使用配体分子量的对数乘以一个缩放因子来近似构象熵损失,尽管这似乎比可旋转键的数量更缺乏机理上的理由。LUDI、Fresno和ChemScore基于可旋转键的数量和每个可旋转键的环境、或其变种,使用特别项来近似配体的熵贡献11。广受欢迎的对接方法AutoDock Vina基于可旋转键数量也使用了一个简单的配体熵罚分来处理打分值12。
因此,使用更现代的对接算法,因其近似考虑了配体的构象熵,应该提高了对虚拟筛选化合物的排序能力。通常会导致非常柔性的化合物在排序中出现的位置比忽略配体熵时出现的位置要低。因此,对于药物设计而言,即使是对柔性配体结合方面的构象熵损失进行粗略估计,也比完全忽略它要好。
机器学习方法
最近机器学习方法取得了巨大进步,这些方法已同步用于对接。在回溯性打分函数比较评估(CASF)试验中,这些方法展现了出色的预测准确性。在某些情况下,这些方法的表现超过了传统基于物理的、对计算需求大得多的基于分子模拟的方法13。原则上,如果使用扼要表述了配体构象熵的特征对配体进行编码,那么比起忽略熵或使用近似方法考虑熵的传统对接算法,能有更优的结合能预测性能。除了改进打分之外,深度神经网络的最新应用还允许进行对接和亲和力计算。例如,Francoeur及其同事14最近报道了一项令人印象深刻的研究,其中使用了3D卷积神经网络来编码配体和受体的特征。这些特征随后用于针对PDB中与多个相似结合口袋对接的2250万个配体模式,预测结合模式和亲和力,并取得了良好的准确度。
分子动力学模拟
对接打分最佳分子的分子动力学模拟,也可以从最近的进展受益,这些进展以计算高效的方式考虑了配体熵。例如,Duan等人报道了一种理论严格、计算高效且数值可靠的方法,用于计算蛋白质-配体结合中熵对自由能的贡献。不同于广泛采用、但计算成本高昂、用于熵变计算的正则模方法,他们的方法从分子动力学模拟直接计算结合自由能的熵成分,无需额外的计算成本15。
D’Aquino等人报道16,熵变可以作为自由分子与结合状态中构象受限分子之间的熵差异来计算。假设具有加和性,他们可以计算较大分子结合时的熵变。他们提供了一个构象熵值的查寻表,该表根据每个二面角相邻的原子类型进行参数化,可用于计算与复杂分子结合相关的熵变。这是一种非常快速的构象熵变化评估方法,适合于快速筛选算法16。
显然,在对接实验和MDS中精确地对配体结合自由能排序时,考虑配体的构象熵是重要的。考虑到从第一性原理计算绝对结合自由能,以及用炼金术法计算准确的相对结合自由能的难度和计算费用,近似和经验校是实现这一目标的最可行方式。正如在量子化学中的计算瓶颈所示,例如,机器学习方法可能在不久的将来,提供一种在计算可行性和准确性之间更好的折中方案。
推荐阅读
参考文献
- Lyu, J.; Wang, S.; Balius, T. E.; Singh, I.; Levit, A.; Moroz, Y. S.; O’Meara, M. J.; Che, T.; Algaa, E.; Tolmachova, K.; Tolmachev, A. A.;Shoichet, B. K.; Roth, B. L.; Irwin, J. J. Ultra-Large Library Docking for Discovering New Chemotypes. Nature 2019, 566, 224−229.
- Mondal, D.; Florian, J.; Warshel, A. Exploring the Effectiveness of Binding Free Energy Calculations. J. Phys. Chem. B 2019, 123, 8910−8915.
- Aldeghi, M.; Heifetz, A.; Bodkin, M. J.; Knapp, S.; Biggin, P. C. Accurate Calculation of the Absolute Free Energy of Binding for Drug Molecules. Chem. Sci. 2016, 7, 207−218.
- Guterres, H.; Im, W. Improving Protein-Ligand Docking Results with High-Throughput Molecular Dynamics Simulations. J. Chem. Inf. Model. 2020, 60, 2189−2198.
- Genheden, S.; Ryde, U. The Mm/Pbsa and Mm/Gbsa Methods to Estimate Ligand-Binding Affinities. Expert Opin. Drug Discovery 2015,10, 449−61.
- Sun, H.; Duan, L.; Chen, F.; Liu, H.; Wang, Z.; Pan, P.; Zhu, F.; Zhang, J. Z. H.; Hou, T. Assessing the Performance of MM/PBSA and MM/GBSA Methods. 7. Entropy Effects on the Performance of End-Point Binding Free Energy Calculation Approaches. Phys. Chem. Chem. Phys. 2018, 20, 14450−14460.
- Udugamasooriya, D. G.; Spaller, M. R. Conformational Constraint in Protein Ligand Design and the Inconsistency of Binding Entropy. Biopolymers 2008, 89, 653−67.
- Chang, C. E.; Chen, W.; Gilson, M. K. Ligand Configurational Entropy and Protein Binding. Proc. Natl. Acad. Sci. U. S. A. 2007, 104,1534−9.
- Chodera, J. D.; Mobley, D. L. Entropy-Enthalpy Compensation: Role and Ramifications in Biomolecular Ligand Recognition and Design. Annu. Rev. Biophys. 2013, 42, 121−42.
- Huang, S. Y.; Zou, X. Inclusion of Solvation and Entropy in the Knowledge-Based Scoring Function for Protein-Ligand Interactions. J. Chem. Inf. Model. 2010, 50, 262−73.
- Guedes, I. A.; Pereira, F. S. S.; Dardenne, L. E. Empirical Scoring Functions for Structure-Based Virtual Screening: Applications, Critical Aspects, and Challenges. Front. Pharmacol. 2018, 9, 1089.
- Trott, O.; Olson, A. J. Autodock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455−461.
- Li, H.; Sze, K. H.; Lu, G.; Ballester, P. J. Machine-Learning Scoring Functions for Structure-Based Drug Lead Optimization. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2020, 10, No. e1465.
- Francoeur, P.; Masuda, T.; Sunseri, J.; Jia, A.; Iovanisci, R. B.; Snyder, I.; Koes, D. R. 3d Convolutional Neural Networks and a Crossdocked Dataset for Structure-Based Drug Design. J. Chem. Inf. Model. 2020, 60, 4200.
- Duan, L.; Liu, X.; Zhang, J. Z. Interaction Entropy: A New Paradigm for Highly Efficient and Reliable Computation of Protein-Ligand Binding Free Energy. J. Am. Chem. Soc. 2016, 138, 5722−8.
- D’Aquino, J. A.; Freire, E.; Amzel, L. M. Binding of Small Organic Molecules to Macromolecular Targets: Evaluation of Conformational Entropy Changes. Proteins: Struct., Funct., Genet. 2000, 41, 93−107.


