
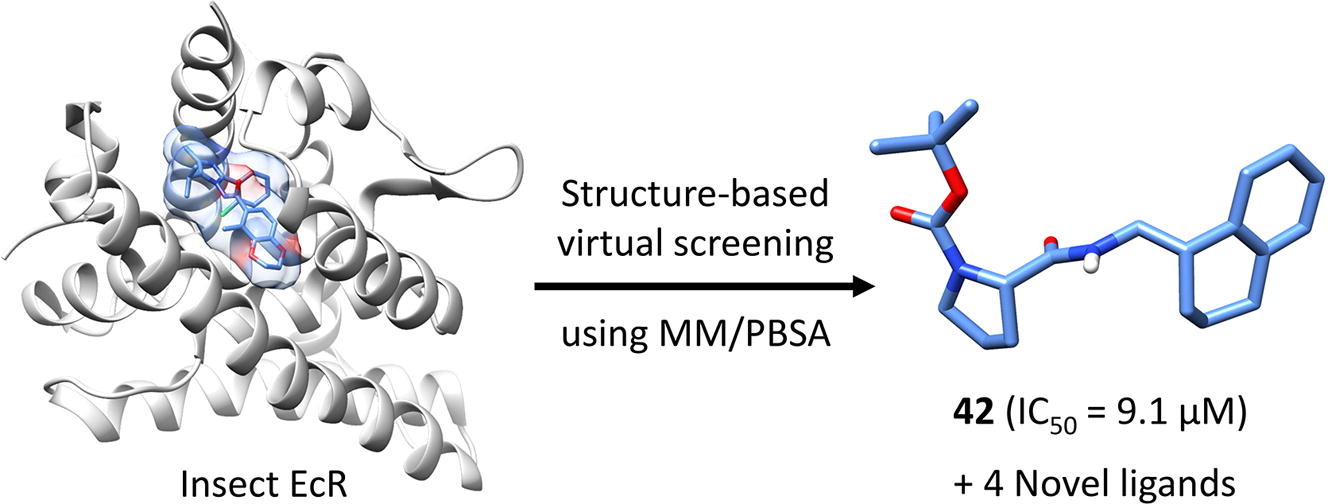
摘要:在本研究中,作者用MM/PBSA方法预测配体的EcR结合活性。首先 使用40个已知EcR配体的验证研究表明,当引入配体的构象自由能项时,令人满意地预测了结合活性。 随后,将该MM/PBSA方法应用于基于结构的分级虚拟筛选,从380万化合物的数据库中挑出12个化合物,其中5个化合物在基于细胞的竞争性结合测试中具有活性。
Horoiwa, S., Yokoi, T., Masumoto, S., Minami, S., Ishizuka, C., Kishikawa, H., … Miyagawa, H. (2019). Structure-based virtual screening for insect ecdysone receptor ligands using MM/PBSA. Bioorganic & Medicinal Chemistry, 27(6), 1065–1075. https://doi.org/10.1016/j.bmc.2019.02.011
前言
蜕皮激素受体(EcR)是一种昆虫核受体,由蜕皮激素20-羟基蜕皮激素激活。 由于合成的EcR配体会破坏昆虫的正常生长,因此它们是有吸引力的新型杀虫剂候选物。 在本研究中,作者用MM/PBSA方法预测配体的EcR结合活性。 使用40个已知EcR配体的验证研究表明,当引入配体的构象自由能项时,令人满意地预测了结合活性。 随后,将该MM/PBSA方法应用于基于结构的分级虚拟筛选,从380万化合物的数据库中挑出12个化合物,其中5个化合物在基于细胞的竞争性结合测试中具有活性。最有效的化合物是简单的脯氨酸衍生物,具有低微摩尔结合活性,是有价值的先导化合物,有待进一步结构优化。
MM/PBSA方法的验证
作者首先用了40个已知的EcR配体进行方法验证研究:探讨MM/PBSA能否有足够的精度预测40个配体的活性。过程详述如下:
验证用配体数据集
13个化合物由合成而得,15个化合物由Mitsui Chemicals Agro公司友情赠送,12个化合物来自自家的库存样品,化合物的结构与活性见原文表1.
EcR的同源建模
e S. frugiperda EcR (SfEcR) 配体结合域(LBD)的3D结构用Modeller 9.16同源建模而得。SfEcR LBD一级序列用GenBank获取号AAM55494与 CAD58232的两段部分序列拼合而来。模板用了HvEcR LDB与ecdusterioid EcR晶体结构,PDB code分别为3IXP与1R1K。
分子对接
OpenEye的OEDocking 3.2.0.2软件包用于分子对接计算。EcR结合位点的模型用MAKE_RECEPTOR模块生成;化合物的构象系综用OMEGA 2.5.1.4准备;分子对接用HYBRID进行计算。
分子动力学模拟与MM/PBSA计算的准备
AMBER 16软件包用与分子动力学模拟(MD Simulation,MDS)研究以及MM/PBSA计算。AMBER 16的TLEAP模块用来准备MDS的模拟体系。蛋白-配体复合物结构由上一步的分子对接而来。原子电荷用内置于antechamber模块的AM1-BCC法计算而得,蛋白采用AMBER force field 14SB(ff14SB)力场,配体采用general AMBER force field 2(GAFF2)力场。体系用Na+与Cl–离子进行电荷中和并包于由TIP3P水分子模型组成的10Å 厚的立方体水盒子中。
分子动力学模拟
AMBER 16的pmemd.cuda模块用来进行MDS计算,所有的体系都用了周期边界条件。Particle mesh Ewald(PME)计算长程静电相互作用截断值为12.0Å。MDS的时间步长(time step)设为1.0fs。共价连接的氢原子采用SHAKE算法进行了约束。在MDS之前,体系先进性最陡下降法与共轭梯度法进行能量最小化以获得小于0.5 × 10−4kcal mol−1 Å−2的梯度。然后将体系在恒容系综里(NVT)逐步在20ps里用0.5kcal mol−1 Å−2的约束从100K升温至300K。然后在常压系综(NPT)中进行180ps的平衡。生产计算(Production run)在NPT系综进行150ps模拟,蛋白-配体复合物坐标每1ps保存此意用于MM/PBSA计算用。对于每个配体分别进行3次不同初始速度的分子动力学模拟,最后每个配体总共得到450(150×3)帧快照。
MM/PBSA计算
蛋白-配体结合自由能(ΔGbind)可以表示Eq 1的三个态的自由能差值:
ΔGbind = GPL -GP – GL (1)
其中下标PL, P与L分别代表蛋白-配体复合物,Apo蛋白(不结合配体的蛋白)与游离的配体。在MM/PBSA中,每个态的自由能用Eq 2来评估:
ΔGX = EMM + GPB + GSA -TS (2)
其中下标X代表PL,P与L。在上述方程中,EMM是气相中分子力学能量,为库伦相互作用能、范德华相互作用能与键能的总和。GPB是溶剂化自由能的极性相互项,用PB(Poisson-Boltzmann)方程计算;GSA是溶剂化自由能的非极性项,用溶剂可及表面积来预估;TS是溶质的熵乘以绝对温度。在本文中,仅配体的构象自由能被加以考虑(见下文)。复合物结构MDS轨迹作为Apo蛋白与游离配体的轨迹来源。MM/PBSA用AMBER 16的MMPBSA.py.MPI模块来进行计算。内部与外部的介电常数分别设为1.0与80.0。
配体构象自由能计算
配体构象自由能(Ligand conformational free energy)用OpenEye SZYBKI 1.9.03的FreeForm模型进行计算。FreeForm内置了快速的构象熵计算方法。MM/PBSA用每化合物450帧的构象进行计算,为了减少计算量,配体的构象自由能用每化合物45帧构象进行计算。
结果
作者考察了三个MM/PBSA计算策略(A,B,C):
- 策略A:Single-point MM/PBSA
- 策略B:MD-based MM/PBSA
- 策略C:MD-based MM/PBSA + ΔGconf_ligand
对复合物结构进行MM最小化获得的单一结构进行MM/PBSA打分。
基于复合物结构MDS轨迹进行MM/PBSA打分。
在策略B基础上考虑了配体的构象自由能:MD-based MM/PBSA + ΔGconf_ligand。
出于比较的目的,也考察了对接打分值与实验值的关系。三种MM/PBSA预测结合自由能,分子对接打分值与实验pIC50的关系如图1所示。其中实线为回归趋势线,R值为Pearson相关性系数。
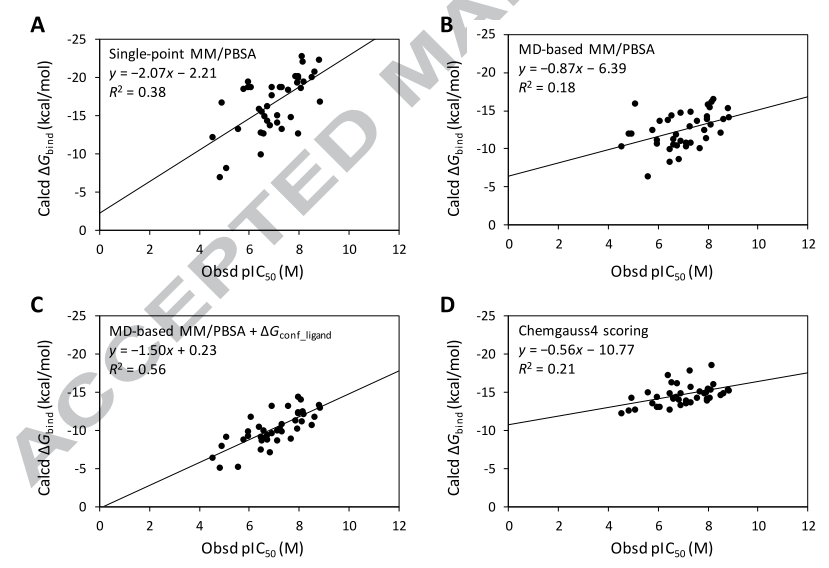
Figure 1. 实验值pIC50与计算的结合自由能之间的关系:(A)single-point MM/PBSA;(B)MD-based MM/PBSA;(C)MD-based MM/PBSA+ΔGconf_ligand;(D)对接打分值(Chemgauss4 score)。其中实线为回归趋势线,R值为Pearson相关性系数。
如图1 3B所示,MD-based MM/PBSA方法与实验结合亲和力之间的一致性很差(R2=0.18)。令人惊讶的是,这甚至比分子对接打分值与实验值之间的相关性(R2=0.21)还差。然而,如图1 2C所示,引入了配体构象自由能(ΔGconf_ligand)之后,显著且巨大地提升了相关性(R2=0.56)。
值得注意的是,图1 2C的回归线与截距是相当合理的。尽管40个受试的配体具有结构上的相似性,但是它们构象自由能具有相当大的差别,分布范围从0.44到6.9kcal/mol。ΔGconf_ligand项的巨大贡献主要是因为有些配体在结合位点里被迫摆出的不合理的构象所致。以化合物7与35对接构象为例,它们有相似的骨架、在结合位点里摆出同样的构象(Figure 2), 然而它们之间的ΔGconf_ligand差值达到了2.84 kcal/mol,这很好地解释了为什么化合物7的活性是35的8500倍。
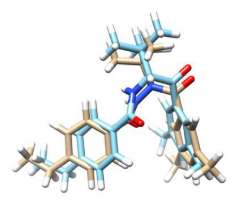
Figure 2. 化合物7与35对接构象的比较
MM/PBSA的构象自由能经常采用游离配体的MDS轨迹用正则模分析(nomal mode analysis,MNA)计算而得。然而,NMA用于大规模虚拟筛选是个挑战,主要是因为一方面收敛具有难度,另一方面计算费用非常昂贵。在本文中,FreeForm用来计算配体的构象自由能。在FreeForm中,构象自由能用OMEGA计算高辩构象系综以及Wlodek等人开发的构象熵算法获得。因此,FreeForm不会有收敛上的问题,并且可以大规模的进行计算,适用于虚拟筛选的场合。
小结
总的来说,MM/PBSA验证结果表明不同策略的预测性能从高到低排序如下:MD-based MM/PBSA+ΔGconf_ligand, Single point MM/PBSA,docking scoring,MD-based MM/PBSA。计算效率排序如下:docking scoring, single point MM/PBSA, MD-based MM/PBSA, MD-based MM/PBSA + ΔGconf_ligand。因此,作者决定采用docking scoring, single point MM/PBSA, MD-based MM/PBSA + ΔGconf_ligand用于分级虚拟筛选。
基于结构的虚拟筛选
虚拟筛选流程如Figure 3所示,分级策略用于筛选含380万化合物的数据库。首先用FRED对接方法进行虚拟筛选,Chemgauss4为打分函数,保留5000个打分最高的化合物;接着,用single point MM/PBSA策略对5000个化合物进行打分,取打分值优于-10kcal/mol的化合物,最后得到打分最高的389个化合物;这389个化合物进一步用MD-based MM/PBSA + ΔGconf_ligand策略进行打分,选择其中ΔGbind≤−9 kcal/mol的化合物并进一步依据结构新颖性与商业可购买性进行了人工挑选。最后12个在结构上与现有的EcR配体截然不同化合物从Namiki Shoji公司采购回来,并进行了活性测试。虽然虚拟筛选用的化合物是立体化学区分的,但是采购回来的化合物是立体化学混合物。
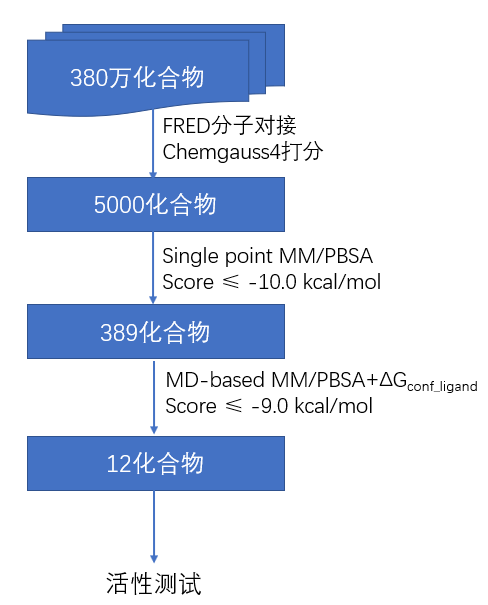
Figure 3. 虚拟筛选流程
12个化合物的纯度均大于90%,放射性配体竞争实验的生物活性测试结果见表1。尽管7个化合物在最高浓度(100或250ΜM)没有表现出结合能力,但是其它的5个化合物(41,42,46,48,50)抑制了放射性配体的结合。最强效化合物42的IC50为9.1uM,大约比上市商品化合物Tebufenozide(化合物7)与Chromafenozide(12)活性弱5000倍。然而,因为化合物42是简单的脯氨酸衍生物,因此它是一个值得进一步优化的起始化合物。
Table 1. 12个化合物的结构、计算的ΔG与实验IC50
| ID | Structure | ΔG | pIC50 | ID | Structure | ΔG | pIC50 |
|---|---|---|---|---|---|---|---|
| 41 |
|
-9.13 | 4.28 | 47 |
|
-12.99 | 小于3.60 |
| 42 |
|
-9.49 | 5.04 | 48 |
|
-9.61 | 4.11 |
| 43 |
|
-14.01 | 小于3.06 | 49 |
|
-10.70 | 小于3.60 |
| 44 |
|
-9.57 | 小于4.00 | 50 |
|
-11.01 | 4.12 |
| 45 |
|
-9.44 | 小于4.00 | 51 |
|
-10.16 | 小于3.60 |
| 46 |
|
-10.73 | 4.13 | 52 |
|
-10.51 | 小于3.60 |
总结
在本研究中,作者将基于MD轨迹的MM/PBSA与FreeForm的构象自由能组合开发了EcR配体结合亲和力预测的高效计算方法。该计算方法用于基于结构的分级虚拟筛选,成功地从380万化合物数据库中识别出5个全新的EcR配体(IC50小于100uM),命中率(42.7%)显著地优于已有文献报道的虚拟筛选方法,这说明该MM/PBSA方法的有效性。
本研究的亮点
- 开发了MD-based MM/PBSA + ΔGconf_ligand结合自由能预测策略;
- 用OpenEye/Fred分子对接进行第一轮的虚拟筛选过滤;
- 用单点MM/PBSA预测结合亲和力,进行了第二轮的过滤;
- MD-based MM/PBSA与构象自由能组合,进行了第三轮过滤。


