
摘要:喹唑啉酮是选择性PI3Kγ与PI3Kδ抑制剂的公共片段,在本研究中和记黄埔的科研人员用片段替换软件BROOD成功地识别出喹唑啉酮的新型替代物吡咯并三嗪酮。在分子对接的指导下,使用新的环状连接臂将该新型特异性片段与抑制剂的铰链结合区连接。进一步针对铰链区进行了SAR优化发现了候选化合物26,它是一种高效、选择性的PI3Kγ/PI3Kδ双重抑制剂,在临床前物种中具有有利的DMPK特性。
原文:Jia, H. et al. (2019) “Discovery, Optimization, and Evaluation of Potent and Highly Selective PI3Kγ–PI3Kδ Dual Inhibitors,” Journal of Medicinal Chemistry, 62(10), pp. 4936–4948. Available at: https://doi.org/10.1021/acs.jmedchem.8b02014.
PI3K与类风湿性关节炎
PI3K激酶(PI3-Kinase或PI3K)是一类酶家族,它们与多种细胞功能相关,比如细胞生长、增殖、分化、运动性、存活和细胞内运输等等。根据序列同源性与脂底物的专一性,PI3K激酶家族分为I,II,III类。其中第I类家族受到广泛的药物开发研究。PI3K I类成员都是杂二聚体,包含一个小的调节结构域与一个大的110kDa催化结构域。催化结构域有4种不同的亚型:p110α,p110β, p110γ与p110δ。
其中PI3Kα与PI3Kβ广泛表达,在细胞的生长、存活与增值中起重要作用,抑制PI3Kα与PI3Kβ主要靶向癌症治疗。PI3Kγ主要表达于粒细胞、单核细胞和巨噬细胞,而PI3Kδ还见于B细胞和T细胞。PI3Kγ或PI3Kδ基因敲除的小鼠存活、可生育,但表现出先天免疫以及适应性应答的显著缺陷。因此,PI3Kγ或PI3Kδ专一的抑制剂是潜在自身免疫疾病的治疗靶标,并且对总的PI3K信号通路不产生干扰、不影响其它细胞的正常功能。
类风湿性关节炎(Rheumatoid arthritis, RA)是一种引起自身免疫疾病,引发关节炎症与组织损伤。T细胞、B细胞、肥大细胞、巨噬细胞和中性粒细胞的活性可以影响疾病的一个或几个方面。考虑到PI3Kδ与PI3Kγ在这些细胞中的功能,以及它们在RA滑膜和成纤维细胞样滑膜细胞(FLSs)中的大量表达,PI3Kδ与PI3Kγ是潜在的类风湿性关节炎治疗靶标。
目前已有PI3Kδ抑制剂进入临床开发阶段。Idelalisib也称为GS-1101、CAL-101或Zydelig(见Figure 1的化合物1),是首个被FDA批准的PI3Kδ抑制剂,用于治疗滤泡性淋巴瘤(FL)和小淋巴细胞淋巴瘤(SLL);与抗CD20利妥昔单抗联合治疗复发性慢性淋巴细胞白血病(CLL)。Umbralisib(见Figure 1的化合物2)是一个小分子PI3Kδ抑制剂,由TG Therapeutics开发,目前处于PIII阶段与抗CD20抗体ublituximab联合治疗血液系统恶性肿瘤。 近年来,越来越多的选择性更好的PI3Kδ抑制剂进入临床试验阶段。
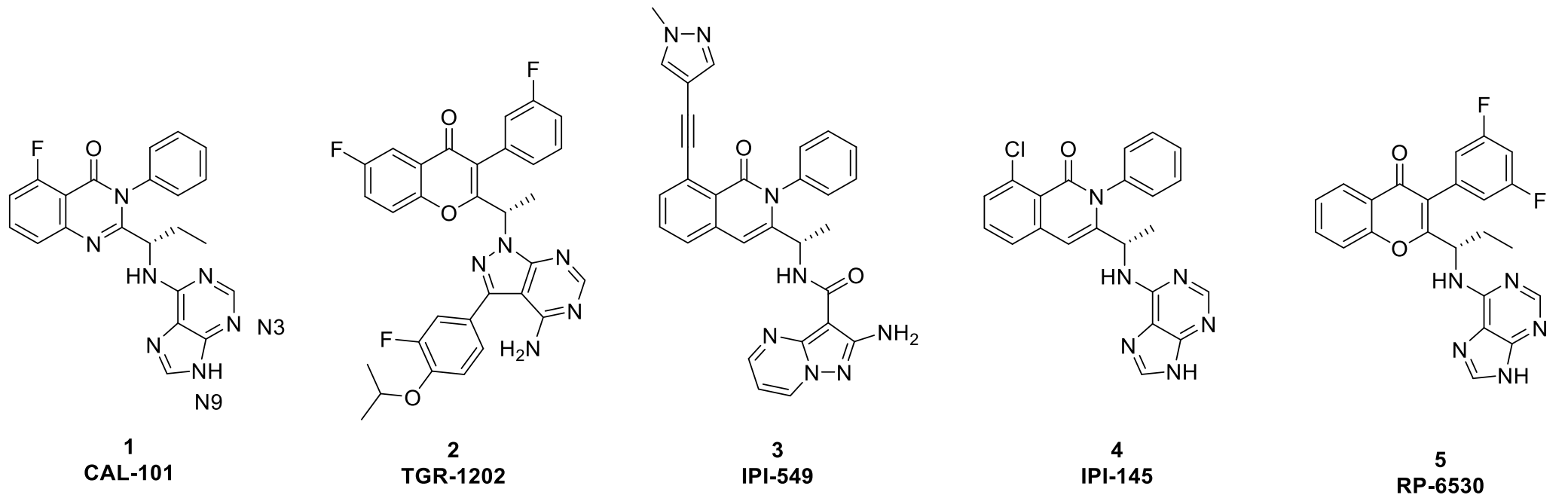
Figure 1. PI3Kγ与PI3Kδ抑制剂
尽管单独抑制PI3Kγ或PI3Kδ在自身免疫和肿瘤疾病中提供了独特的治疗机会,但同时抑制这两种亚型可以在治疗多种复杂的免疫介导的炎性疾病中产生优异的临床功效。 已知这两种酶都有助于支持恶性B细胞和T细胞的生长和存活,在支持性肿瘤微环境的形成和维持中起到关键作用。靶向PI3Kγ和PI3Kδ都可以用于肿瘤治疗,如慢性淋巴细胞白血病,急性髓性白血病,非霍奇金淋巴瘤和多发性骨髓瘤。
Infinity制药公司开发的Duvelisib(见Figure 1化合物4)是首个报道的PI3Kγ/PI3Kδ双重抑制剂,证明通过双重阻断可以实现适应性和先天免疫功能的联合抑制,从而在动物模型中的多种炎症和自身免疫疾病中产生显着的治疗优势。然而,在使用甲氨蝶呤背景的双盲,安慰剂对照的II临床期研究中,Duvelisib未能在RA患者中显示出积极的临床反应。基于作者自己的PKPD模型,他们推测临床剂量(1 mg和5 mg,每天两次)都不能提供足够的药物暴露来抑制这两个靶标(特别是PI3Kγ)以产生临床反应。在2018年,Duvelisib被批准用于治疗患有复发或难治性慢性淋巴细胞白血病/小淋巴细胞淋巴瘤和滤泡性淋巴瘤的患者。 Rhizen Pharmaceuticals S.A.也完成了PI3Kγ/PI3Kδ双重抑制剂Tenalisib(Figure 1化合物5)的I期临床研究,以治疗复发或难治性血液系统恶性肿瘤患者,包括T细胞淋巴瘤。
在本研究中,作者描述了发现新型强效、高选择性PI3Kγ/PI3Kδ双重抑制剂的过程,最后发现了口服有效的、用于治疗RA和其他自身免疫疾病的候选药物26。
用BROOD进行骨架跃迁:改善DMPK
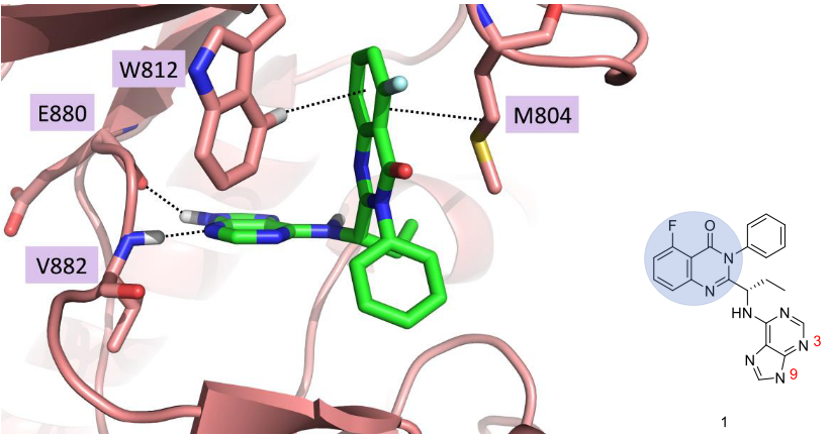
Figure 2. Duvelisib衍生物(化合物1)与PI3Kγ的结合模式(PDB 2CHW):铰链区与V882和E880的骨架形成氢键相互作用;特异性片段与M804发生疏水相互作用,并且与W812有边缘-π相互作用,W812是两种相互作用中较强的一种。
如前面提到,模拟显示Duvelisib(化合物1)在临床上没有对RA显效可能是因为临床剂量(1 mg和5 mg,每天两次)都不能提供足够的药物暴露来抑制这两个靶标(特别是PI3Kγ)以产生临床反应。因此,本项目的首要目标替换化合物1喹唑啉酮片段(Figure 2化合物1阴影区)以发现DMPK性质更理想的化合物。理想的替换片段不仅与化合物1具有相似的匹配,而且具有相似的相互作用模式,尤其是与P-Loop区W812的相互作用。
第一步是使用生物电子等排物替换工具BROOD对分子的喹唑啉酮片段进行虚拟筛选以发现可行的特异性替换片段。BROOD提供了先导物跃迁和头脑风暴式的可选方法,给出许多的潜在片段替代方案。为了对BROOD给出的结果进行合成优先性排序,又用静电相似性工具EON对BROOD结果进行打分,发现了多种潜在的片段。用裸眼比较替代片段与参比片段的静电势,进行可视化分析后,认为吡咯并三嗪酮与化合物1的喹唑啉酮具有最相似的静电特征(见Figure 3),因此选择这个片段进行后续的化学研究。在该研究中作者详细描述了吡咯并三嗪酮PI3Kγ/δ双重抑制剂先导化合物优化的详细过程。
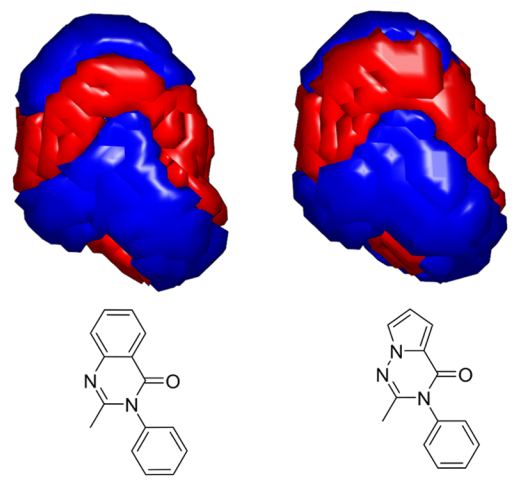
Figure 3. 喹唑啉酮与吡咯并三嗪酮的静电表面比较
开链氨基连接臂的关环:锁定构象
Figure 4. PI3Kγ与PI3Kδ抑制剂的理性设计:开链氨基连接臂环合为五元吡咯烷。
化合物1的结合模式(Figure 2)表明开链氨基连接臂将特异性片段与铰链彼此垂直放置,产生螺旋桨状形状。鉴于分子的整体取向及其在结合口袋内的相互作用,作者意识到将开链氨基连接臂环化成五元吡咯烷将是结构修饰的可行方法,通过锁定构象来增强参数(Figure 4)。初始化合物6(以及随后的7)与PI3Kγ对接计算进一步证实了该想法,对接计算结果显示:在铰链和特异性片段区域中的关键相互作用和化合物整体在结合口袋中的匹配性(Figure 5)与化合物1相当,对接的结合模式还显示:新的环胺连接臂比开链的氨基连接臂更加更充分地占据由M804,I963和M953组成的疏水袋。
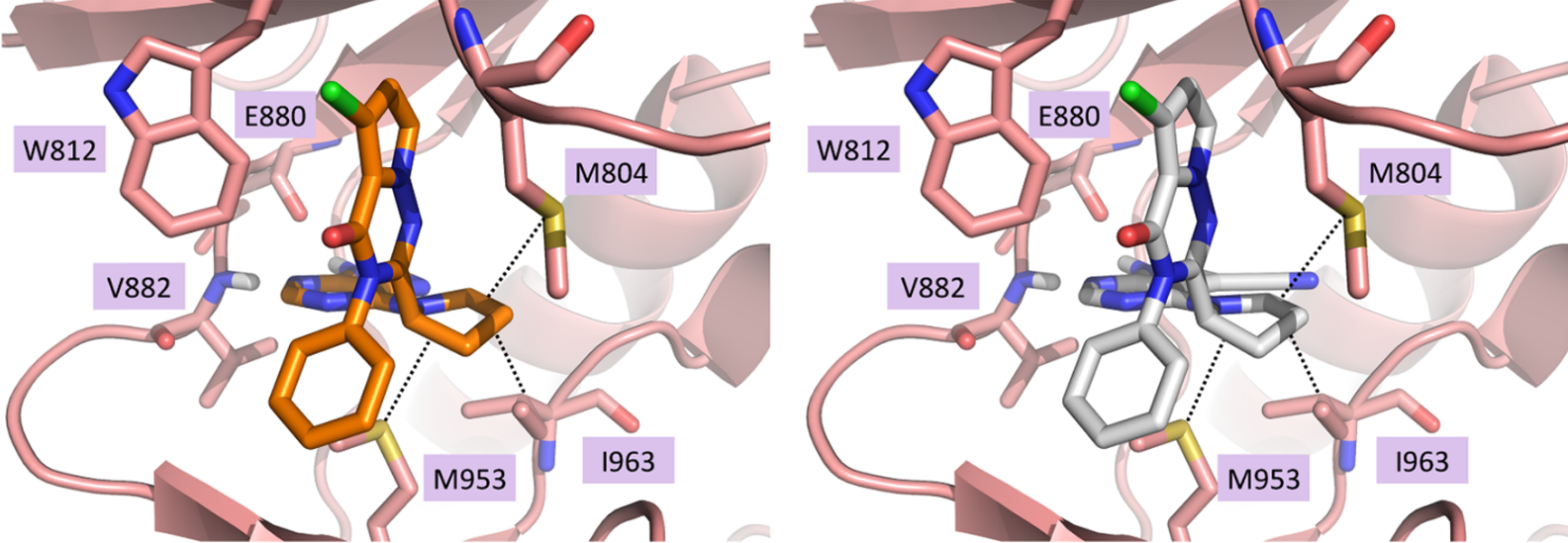
Figure 5. 化合物6、7与PI3Kγ的结合模式。疏水相互作用发生于M804、I963与M953形成一个小的疏水口袋(PDB code:2CHW)。
生物活性测试结果表明上述的假设是对的:化合物6对PI3Kγ与PI3Kδ都表现出良好的抑制活性,IC50分别为0.012μM与0.002μM。与原型的化合物1相比,化合物6保留了对PI3Kδ的活性水平,而提高了对PI3Kγ的抑制活性。化合物7对PI3Kγ与PI3Kδ的IC50分别为0.004μM与0.001μM,提高了对PI3Kγ的抑制活性与化合物4相当。这些数据进一步证实了分子对接的结合模式是对的,并且证实将喹唑啉酮替换为吡咯并三嗪酮、以及将开链氨基连接臂替换为五元吡咯烷环连接臂是正确的。
五元吡咯环的修饰:代谢稳定性的改进
不幸的是,化合物6、7在大鼠的肝微粒体实验中显示出代谢稳定性不好,同时也有必要进一步该进其对PI3Kβ的选择性。在随后SAR研究中,主要以提高选择性,并监控在大鼠与人肝微粒体的代谢稳定性。其中化合物12的对PI3Kβ的选择性提高了50倍,但在大鼠与人的肝微粒体测试实验中还是表现出代谢稳定很差,并且预测被啮齿类动物快速清除。

Figure 6. 化合物12在人与小鼠肝微粒体中的体外代谢物产物
进一步对化合物12的PK与体外代谢产物研究发现:吡咯烷连接臂被氧化是这些化合物的主要代谢途径,见Figure 6。为了减少疏水性吡咯烷的氧化来增加代谢稳定性,引入了4元环氮杂环丁烷作为连接臂(图7),其减少了clogP并保留了活性和选择性所必需的螺旋桨构象。正如所料,与相应的吡咯烷化合物相比,氮杂环丁烷化合物的代谢稳定性得到改善。例如,大鼠肝微粒体实现中氮杂环丁烷20,21和22的剩余百分比分别为63.9%,77.2%和77.5%,并且与相应的吡咯烷类似物7,14和18相比得到很大改善。这种代谢稳定性增加的趋势在人肝微粒体稳定性测试中也很明显(图7)。
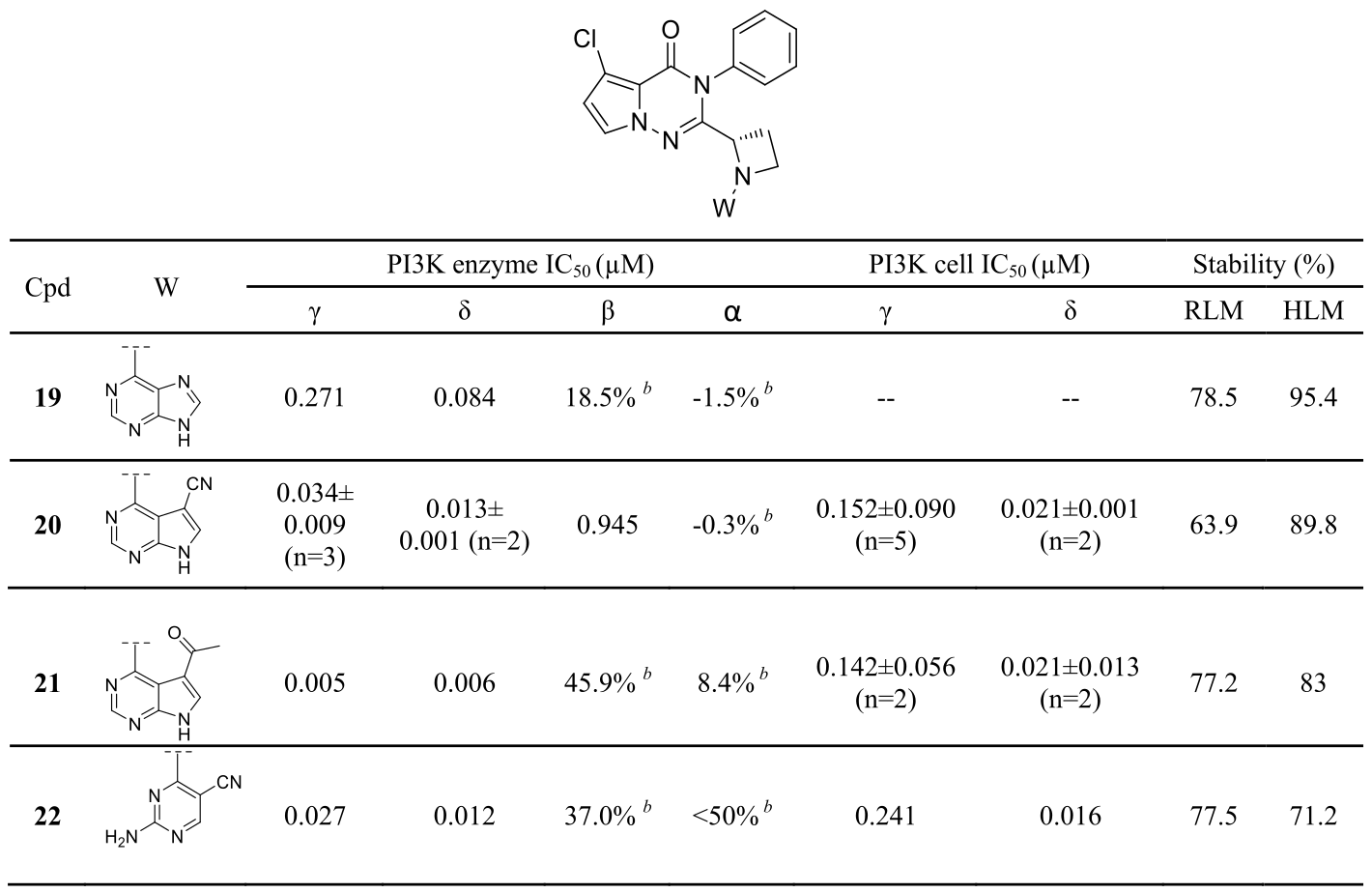
Figure 7. 氮杂环丁烷连接臂化合物的SAR
候选化合物26
对4元环类化合物的铰链结合片段进行SAR研究与优化,如图8所示,最终发现了化合物26。体外ADME筛选表明,化合物26更加稳定,RLM与HLM分别63.5%与81.5%。化合物26显示出体外强效的活性与选择性,对PI3Kγ、PI3Kδ与PI3Kβ的IC50分别为0.004,0.005,0.590μM。在对458个激酶(395个为野生型激酶,63个为突变型激酶)的激酶谱筛选中,在10μM浓度下对PI3K家族具有显著的专一性。此外,对50个GPCR蛋白、离子通道与转运体蛋白均没有活性。
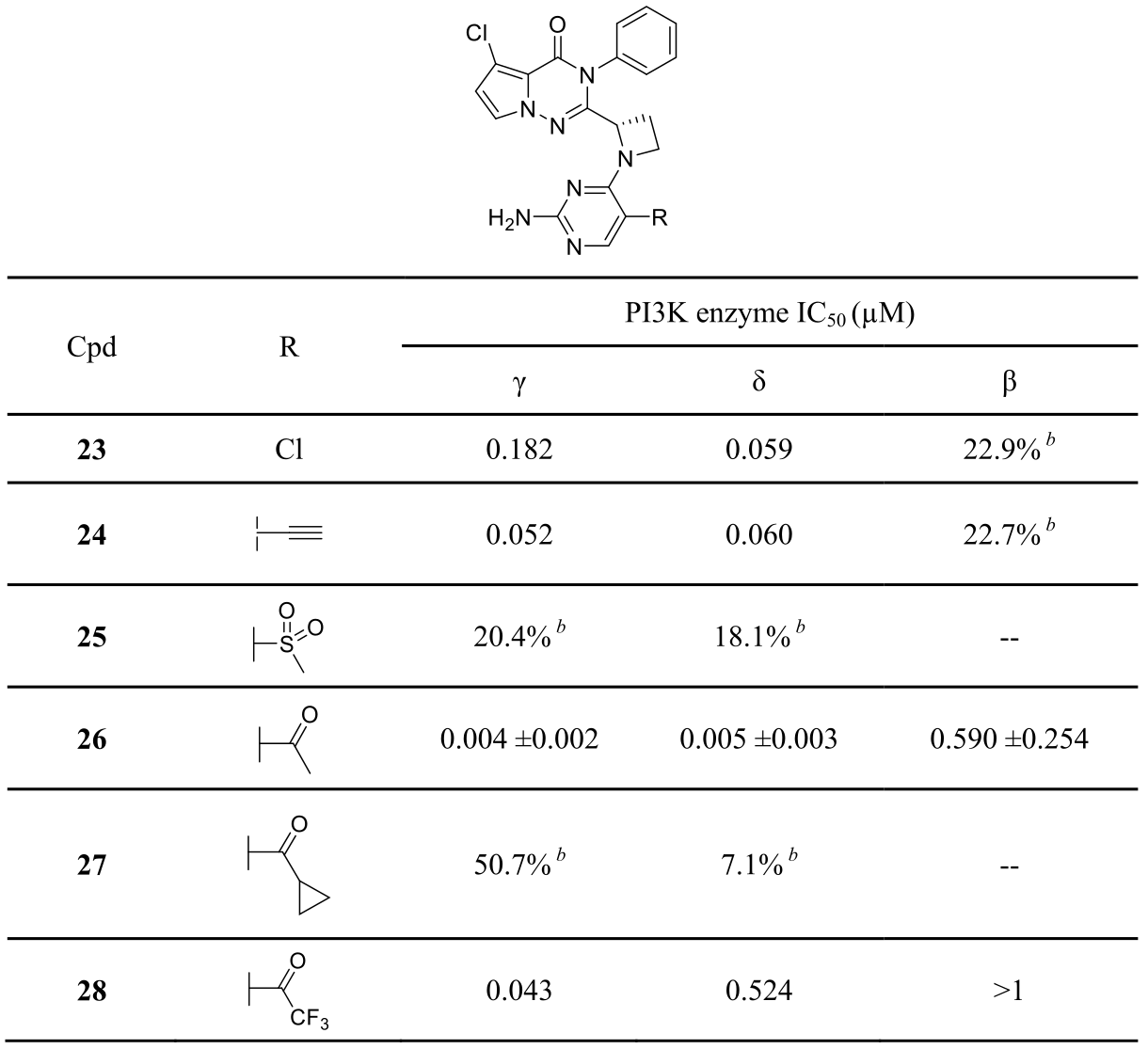
Figure 8. 铰链结合片段的SAR研究与优化
进一步对化合物26进行的体外细胞试验,以了解其对PI3Kδ和PI3K功能活性的影响。PI3Kδ功能的测定包括在人全血中B细胞上的IgM抗体刺激的CD69表达; 大鼠全血B细胞上IgD抗体刺激的CD86表达; 和人全血中fMLP诱导的嗜碱性粒细胞的CD63表达。用于评估PI3Kγ活性的功能测试包括IgE抗体诱导的人全血中嗜碱性粒细胞上的CD63表达。如表4所示,26对嗜碱性粒细胞和B细胞活化表现出有效的抑制作用。 例如,对人全血中嗜碱性粒细胞活化的抑制,IC50为0.042μM和0.337μM,对大鼠全血中B细胞活化的IC50为0.044μM。
用雌性Wistar大鼠进一步评估了化合物26对离体抗IgD诱导的B细胞活化的抑制作用。 26的口服给药在2h有效抑制抗IgD诱导的B细胞活化,在1mg / kg的剂量下抑制88.3%。 将KC / GRO(也称为CXCL1,CXC趋化因子)滴注到大鼠皮下空气囊中诱导了强烈的中性粒细胞趋化性,并且已知该反应涉及PI3K信号传导。 在大鼠的空气袋研究中,26小时阻断KC / GRO诱导的中性粒细胞迁移,在10mg / kg的剂量下抑制87.9%,证实了26对体内PI3Kγ的有效抑制活性。
Table 1. 化合物26的在SD大鼠与Beagle犬上的PK特性

体内给药的药代动力学分析表明,化合物26在大鼠中表现出中度清除率和在狗中表现出低清除率,中等程度的分布容积和高的口服生物利用度(Tabel 1)。当GSH作为辅因子共培养时,在人肝微粒体中未鉴定出谷胱甘肽(GSH)加合物。
在已建立的大鼠胶原诱导的关节炎模型中以0.1,1和10mg/kg每天两次给药26天,从第10-16天起持续7天。它以剂量依赖的方式显着降低平均爪子体积,表明显著的抗炎作用; 估计ED50为0.25mg/kg,BID。1.0mg/kg BID给药,爪体积减少与第11天至第16天的阳性对照YiSaiPu相当; 从第11天起,10mg/kg BID给药表现出强效,并且在第16天疾病进展逆转,几乎使动物恢复至正常状态。
总结
Infinity制药公司开发的Duvelisib是首个报道的PI3Kγ/PI3Kδ双重抑制剂,在使用甲氨蝶呤为背景的双盲、安慰剂对照2期研究中,Duvelisib未能在RA患者中显示出积极的临床反应。作者基于自己的PKPD模拟,他们推测临床剂量(1 mg和5 mg,每天两次)都不能提供足够的药物暴露来抑制这两个靶标(特别是PI3Kγ)以产生临床反应。以此假设为基础,作者首先用BROOD对Duvelisib的喹唑啉酮片段进行跃迁,并用EON进行静电相似性筛选,经过裸眼观察,最后选择了吡咯并三嗪酮做为新的骨架;进一步对开链的连接臂用五元吡咯烷环替换以提高构象稳定性,得到化合物6,7。酶学活性测试表明,化合物6、7的选择性与活性增强,这证明了之前的假设是对的;但是化合物6、7存在代谢稳定性问题。进一步的体外代谢产物研究表明,五元吡咯烷被氧化是主要的代谢途径。因此,作者将五元吡咯烷替换为四元的氮杂环丁烷,既保留了化合物的螺旋桨形状,又保留了关键的相互作用特征,随后的先导化合物优化中发现了化合物26,不仅具有强效的PI3Kγ/δ活性以及PI3Kβ的选择性,而且大鼠与人的肝微粒体代谢稳定性良好。
联系我们
想在自己的项目中使用BROOD,ROCS与EON等OpenEye软件进行骨架跃迁,请联系我们获取免费的试用版;你还可以以委托研究(计算服务)的方式使用该软件。
- 电邮:info@molcalx.com
- 电话:020-38261356


