
摘要:需要不需要对线型先导物进行构象约束以获得更好的衍生物?这是药物化学工作者常常面临的一个问题。最直接的做法是合成出化合物,再去用SPR等动力学实验、ITC等热力学实验来确认是否构象约束达到预期效果。但是合成这些化合物通常并非易事。Freeform可以帮你在合成前采用计算方式评估这种构象约束是否值得一做,然后再决定是否去合成。本文介绍了FreeForm的基本原理、与传统方法相比的优势以及应用实例。
作者:肖高铿/2016-07-02
生物活性构象稳定性与药物化学
将一个长链的先导物(lead)进行构象约束后获得的构象约束衍生物(constrained analog)是药物化学里常用来提高化合物活性的手段。然后,并非每个构象约束的衍生物都能达到预期效果,因此对药物化学科研人员而言,就需要在合成化合物前做出决策:是否值得对先导物进行构象约束?
因为构象约束意味着结构改造,通常需要用到成环的反应,很多时候并非易事。比如图1所示,Ezzili(2011)从Fatty Acid Amide Hydrolase(FAAH)抑制剂先导物出发,采用构象约束策略设计了衍生物。
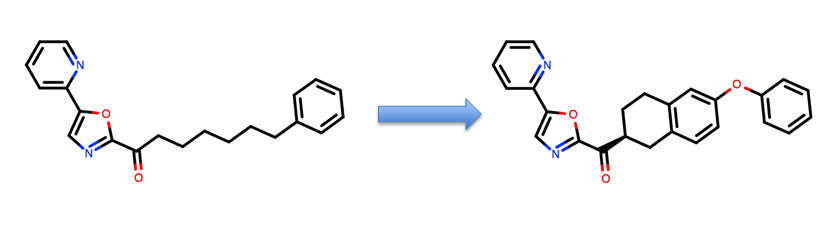
图1. FAAH抑制剂:左边为先导物,右边为构象约束衍生物
因此在合成右边的构象约束衍生物之前需要思考两个问题:
- 柔性的构象应该约束吗?
- 结合构象是否稳定?
化合物结合构象稳定性常采用化合物的构象张力能(Strain Energy)来评价:通常以全局最低能构象为参比,以构象的相对能量值来评估,如图2所示。
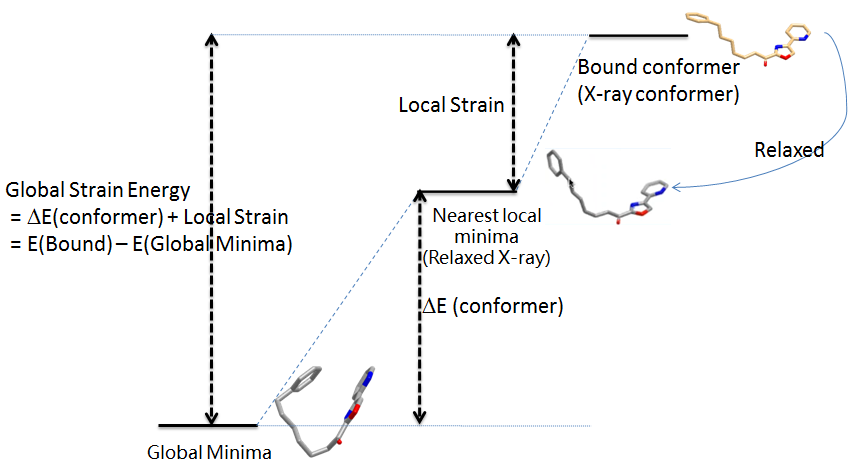
图2. 传统的Strain Energy:以全局极小构象为参比,评估结合构象的稳定性。
传统化合物张力能计算方法存在很明显的问题:只考虑了活性构象与全局极小点,并忽略了熵的变化。而配体分子与靶点结合时真实的情况却是:在溶剂中从非结合的构象系综变为在蛋白结合口袋中一个特定的生物活性构象,如图3所示。
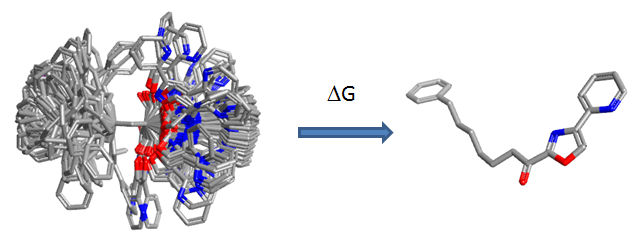
图3. 配体分子与靶点结合的构象变化:从溶剂非结合构象系综变为到生物活性构象
传统的张力能计算虽然直观且容易凭经验用目测即可比较高低,但存在上述的缺陷常常不能正确评估构象稳定性。因此我们需要一个更合适的张力能计算方法。
FreeForm:生物活性构象的张力能计算
FreeForm简介
Freeform是OpenEye药物设计软件包的一个应用,可以用来计算化合物的溶剂化自由能以及计算化合物从一个构象系综变为一个生物活性构象或其它单一构象的自由能变化,如图4所示。
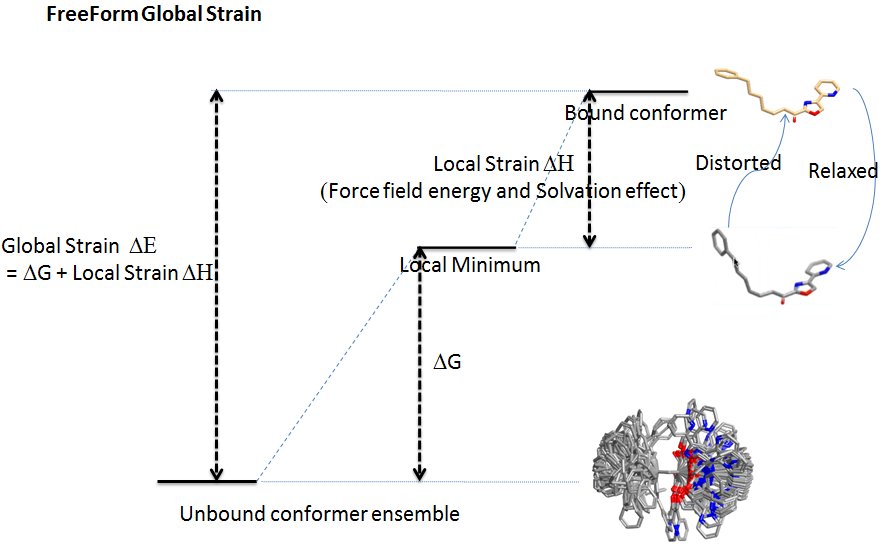
图4. FreeForm计算构象自由能不仅考虑了配体的非结合构象系综,还考虑了主要的熵成分
FreeForm的使用
假设你有一个化合物的生物活性构象:sitagliptin.mol2, freeform可以计算其生物活性构象的全局张力能(Global Strain Energy):
1 2 3 | freeform -calc conf -in sitagliptin.mol2 \ -track sitagliptin.mol2 \ -prefix sitagliptin.3D.pfn |
计算结果会报告在sitagliptin.3D.pfn.pdf的文件里,打开得到类似图5的结果:

图5. sitagliptin.mol2计算结果报告
我们可以看到,对sitagliptin的6个柔性键进行穷尽的构象分析发现了111个极小点构象,其中棕色点为不包含熵贡献的张力能全局极小点,蓝色点为ΔG全局极小点,橘红色点为生物活性构象。我们可以发现,不包含熵贡献的势能全局最小点与ΔG全局最小点不是同一个构象,生物活性构象也并非一个极小点。图5的计算结果报告了全局张力能(Global Strain ΔE)及其各个成分,如表1所示。
表1. sitagliptin的全局张力能(Global ΔE)及其成分
| Items | Values |
| Relaxed Bioactive ΔG : | 2.68 kcal/mol |
| Local Strain ΔH : | 3.02 kcal/mol |
| Global Strain ΔE : | 5.70 kcal/mol |
其中,Global Strain ΔE = Relaxed Bioactive ΔG + Local ΔH
FreeForm的应用
通过比较先导物与衍生物的Global ΔE就可以比较两者的构象稳定性,并进而判断构象约束是否值得做。
FreeForm用于评估构象稳定性案例
案例1. FAAH抑制剂的先导物是否值得进行构像约束?
还是以图1所示的Ezzili的FAAH抑制剂为例,我们分别计算线型的先导物与环约束衍生物的结合构象Global Strain ΔE,结果如图6所示。
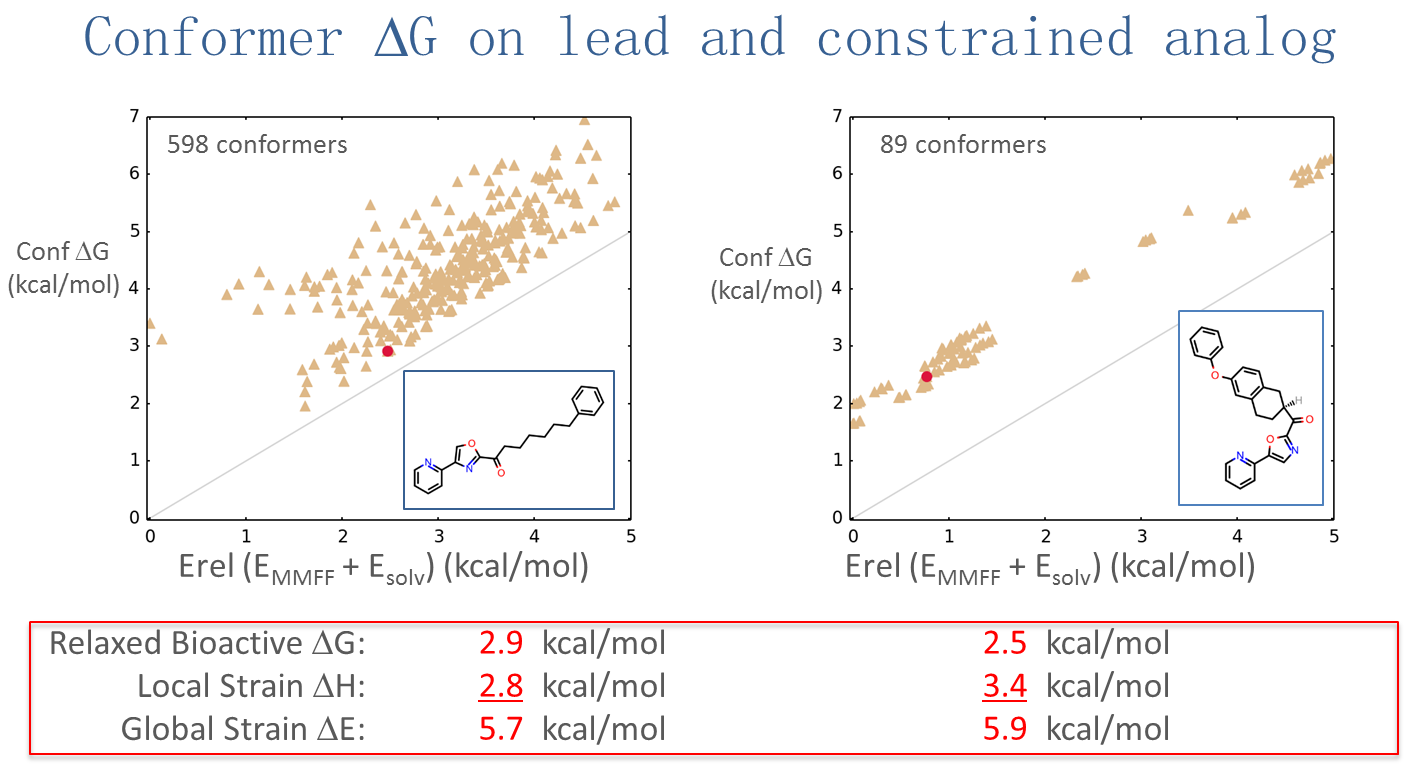
图6. FAAH抑制剂先导物(左边)与构象约束衍生物(右边)的张力能
FAAH抑制剂先导物(左)与构象约束衍生物(右)有相似的Global Strain ΔE, 分别为5.7与5.9 kcal/mol, 构象约束衍生物比之线型的先导物来说没有优势,因此从这一点上讲构象约束衍生物不值得去合成。实际上,线型先导物与构象约束衍生物的Ki值分别为4.7nM与4.4nM(Ezzili, 2011),如表2所示,构象约束衍生物比其先导物并无显著的生物活性优势,与计算结果一致!
表2. FAAH抑制剂先导物与衍生物的构象稳定性评估
| Items | Lead | Constrained analog |
|---|---|---|
| Relaxed Bioactive ΔG | 2.90 kcal/mol | 2.50 kcal/mol |
| Local Strain ΔH | 2.80 kcal/mol | 3.40 kcal/mol |
| Global Strain ΔE | 5.70 kcal/mol | 5.90 kcal/mol |
| Ki | 4.7 nM | 4.2 nM |
案例2. BACE抑制剂的环化
Ellen W. Baxter等人(2007)高通量筛选发现了BACE-1抑制剂,其Ki=900nM。X-ray结果显示发现这类化合物在结合位点折叠为发夹状,如图7所示。
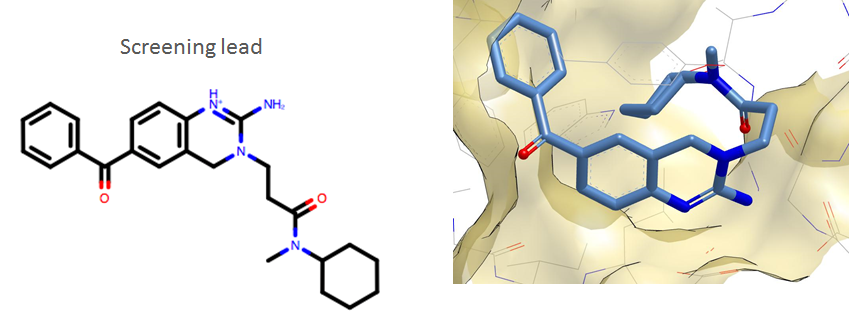
图7. BACE-1抑制剂先导物(左边)及其结合构象(右边)
Huang Yifang等人(2010)从该发夹型结构获得提示:将化合物的两个末端连接起来生成大环结构可以强化发夹型结构,或许提高化合物的活性。为此设计了图8左的大环化合物,结果表明其活性提高到Ki=158nM。我们用FreeForm进行回溯性计算,来看看是否值得去制备这个大环衍生物。
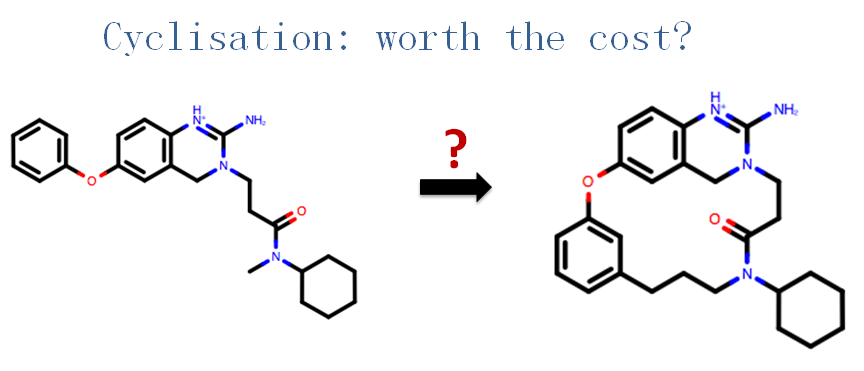
图8. BACE-1抑制剂发夹结构衍生物(左边)及其环化衍生物(右边)
图9总结了先导物与大环化合物的计算结果,我们可以发现:大环分子的构象自由能(relaxed bioactive conformer)稍微不占优势;但是Local Strain ΔH占绝对优势,发夹型化合物需要花能量将化合物折叠起来。总的结果是大环分子的Global Strain ΔE比先导物要低了大约5kcal/mol,即一倍多。由此可以预测:比起先导化合物来,大环化合物的活性有可能提高一倍,值得一试!
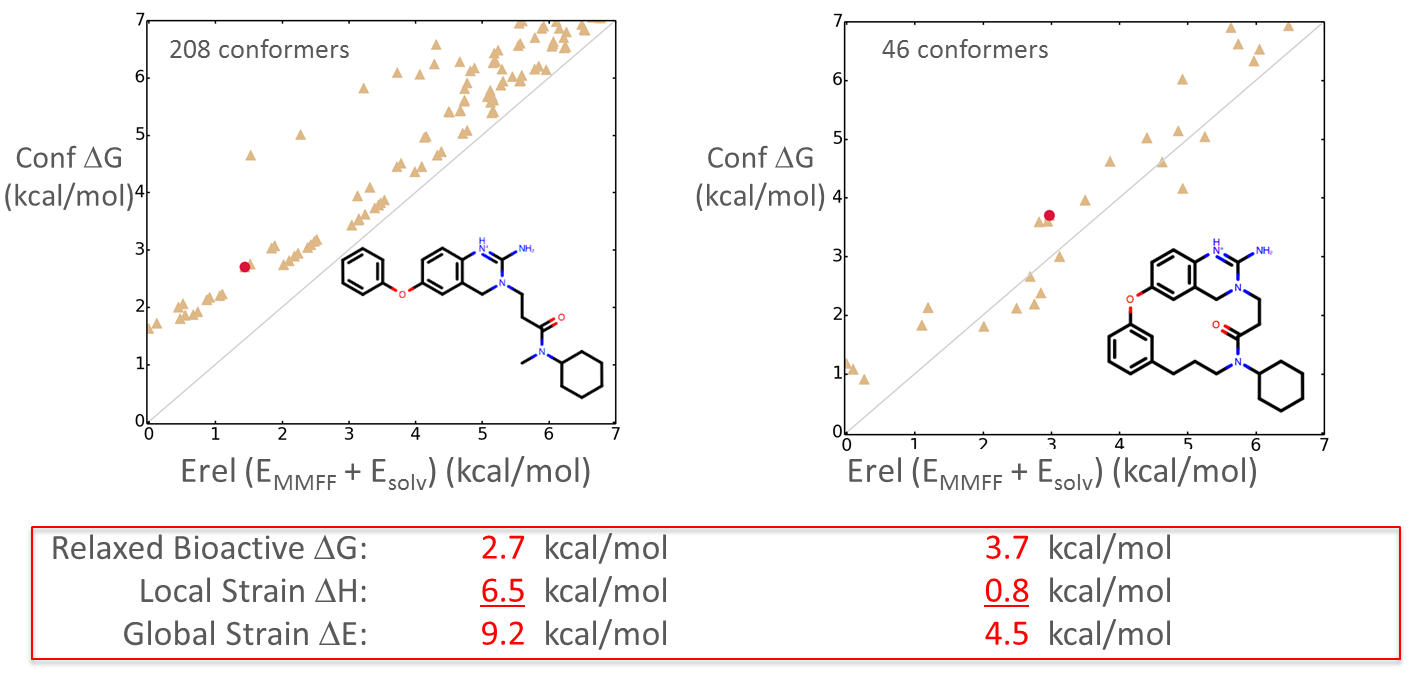
图9. BACE-1抑制剂先导物及其环化衍生物的构象稳定性
实验结果表明,大环衍生物的生物活性要比发夹型先导物高出一倍,大环化合物与发夹型先导物的Ki值分别为60nM(Huang 2010)与158nM(Baxter 2007), 虽然与计算不能定量相关,但是非常一致,如表3所示。
表3. BACE-1抑制剂先导物与大环衍生物的构象稳定性评估
| Items | Lead | Constrained analog |
|---|---|---|
| Relaxed Bioactive ΔG | 2.70 kcal/mol | 3.70 kcal/mol |
| Local Strain ΔH | 6.50 kcal/mol | 0.8 kcal/mol |
| Global Strain ΔE | 9.20 kcal/mol | 4.5 kcal/mol |
| Ki | 158 nM | 60 nM |
案例4. 大环化EGFR激酶抑制剂BI-4020先导化合物5与6的发现与设计
Engelhardt等人(2019)报道了大环激酶抑制剂BI-4020的发现过程,其关键点是:通过筛选激酶抑制剂数据库发现高选择性但中等活性的苯并咪唑类化合物,然后通过大环化将分子完全刚性化。在进行大环化的优化过程中,解释了化合物5、6(见图11)与EGFR激酶的共晶结构,PDB代码分别为6S9C与6S9D。
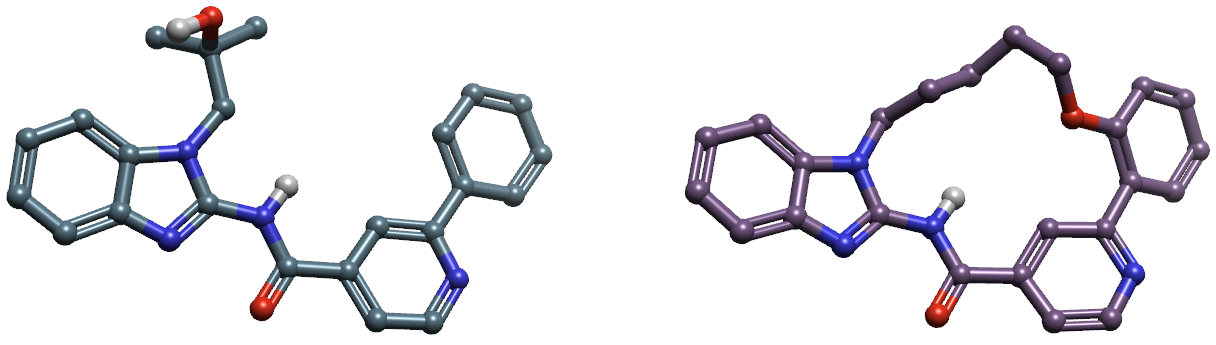
图11. 大环化EGFR激酶抑制剂先导化合物5(左)与6(右)
如前所述,大环化是为了稳定构象使得熵变对结合有利。从共晶结构揭示的相互作用角度看,从化合物5到6的大环化设计并没有增加或减少相互作用,因此它们的活性差异可能是构象张力能差异带来的。分别从PDB 6S9C与6S9D提取了化合物5与6的生物活性构象,用FreeForm计算了全局张力能,结果分别见图12与13。
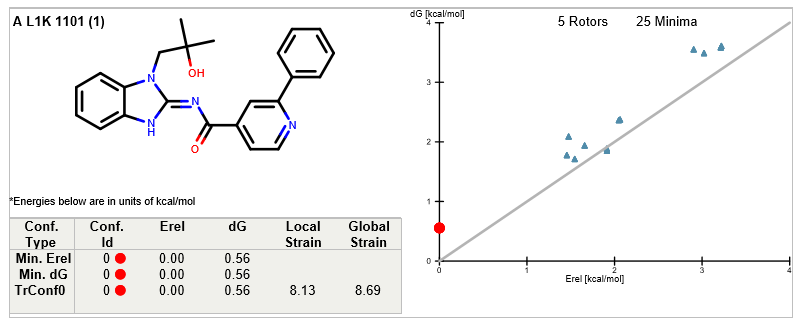
图12. 化合物5的全局张力能计算
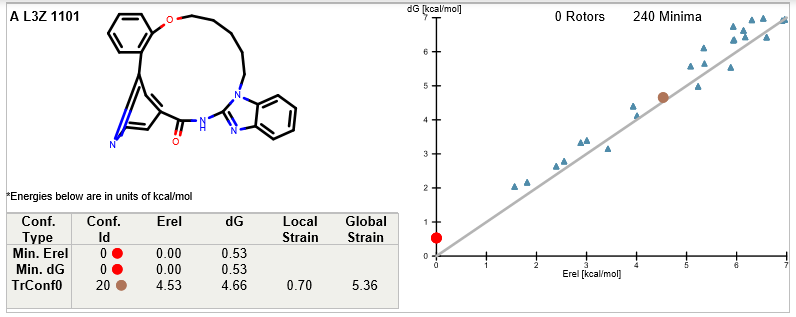
图13. 化合物6的全局张力能计算
化合物5与6的全局张力能计算总结见表5,可以发现开环化合物5比大环化合物6在局部极小点自由能(Relaxed bioactive conformer ΔG)占优势(分别为0.56与4.66kcal/mol);但是在Local Strain ΔH远远地处于弱势(化合物5,6分别为8.13与0.70kcal/mol)。也就是开环化合物5需要比大环化合物6花更大的能量去维持活性构象。这导致化合物5比化合物6具有更高的全局张力能(Global Strain ΔE),分别为8.69与5.36kcal/mol。总的来说,在大环化过程中没有增加或减少相互作用的情况下,全局张力能与化合物的活性的直接相关。从配体的全局张力能角度讲,化合物6比5更有优势,大环化合物6值得一试,这与从酶学实验观察到的IC50一致。
表5. 化合物5与6的全局张力能(Global Strain ΔE)计算结果
| Items | 5 | 6 |
|---|---|---|
| PDB code | 6S9C | 6S9D |
| Relaxed Bioactive ΔG | 0.56 kcal/mol | 4.66 kcal/mol |
| Local Strain ΔH | 8.13 kcal/mol | 0.70 kcal/mol |
| Global Strain ΔE | 8.69 kcal/mol | 5.36 kcal/mol |
| IC50 | 267 nM | 16 nM |
总结
- 传统张力能采用全局最低能构象作为参比,虽然很直观,但忽略了配体熵,这不总是对的;
- 真正的参比应该是溶液中的构象系综,并不直观,需要计算才能知道;
- 构象自由能是对药物化合物科研人员来说是更合理的指标;
- FreeForm的Global Strain ΔE包含了构象自由能的主要成分,为药物化学科研人员提供更好的指导作用;
- 分子对接虚拟筛选的假阳性主要来源之一是构象的能量可及性问题,与构象稳定是一回事,因此也适合于分子对接pose的评估。
推荐阅读
文献
- Baxter, E. W., et al. (2007). “2-Amino-3,4-dihydroquinazolines as Inhibitors of BACE-1 (β-Site APP Cleaving Enzyme): Use of Structure Based Design to Convert a Micromolar Hit into a Nanomolar Lead.” Journal of Medicinal Chemistry 50(18): 4261-4264.
- Huang, Y., et al. (2010). “Macrocyclic BACE inhibitors: Optimization of a micromolar hit to nanomolar leads.” Bioorg Med Chem Lett 20(10): 3158-3160.
- Ezzili, C., et al. (2011). “Reversible Competitive α-Ketoheterocycle Inhibitors of Fatty Acid Amide Hydrolase Containing Additional Conformational Constraints in the Acyl Side Chain: Orally Active, Long-Acting Analgesics.” Journal of Medicinal Chemistry 54(8): 2805-2822. DOI:10.1021/jm101597x
- Wienen-Schmidt, B. et al. (2018) “Paradoxically, Most Flexible Ligand Binds Most Entropy-Favored: Intriguing Impact of Ligand Flexibility and Solvation on Drug-Kinase Binding,” Journal of Medicinal Chemistry, 61(14), pp. 5922–5933. Available at: https://doi.org/10.1021/acs.jmedchem.8b00105.
- Engelhardt, H. et al. (2019) “Start Selective and Rigidify: The Discovery Path toward a Next Generation of EGFR Tyrosine Kinase Inhibitors,” Journal of Medicinal Chemistry, 62(22), pp. 10272–10293. Available at: https://doi.org/10.1021/acs.jmedchem.9b01169.


