
分子内硫-孤对电子相互作用
摘要:本文以一对结构仅一个原子差异但活性有65倍差异的Factor Xa抑制剂为例,分别用GNINA分子对接与Flare QM扭转角分析对结合模式进行打分与扭转角张力能计算,结果表明:不考虑分子内硫-孤对电子相互作用的分子对接打分函数不能区分这对分子的实验活性差异;基于QM的扭转角分析可以捕捉到有利的硫-孤对电子相互作用,发现扭转角张力能差异,并用来解释实验观察到的活性差异。分子内硫-孤对电子相互作用对小分子药物设计以及与受体的相互作用影响不可忽略,Flare QM可以方便地进行此类相互作用研究。
前言
分子内硫-孤对电子相互作用(Sulfur-lone pair interaction)对于小分子药物设计以及对小分子与受体的相互作用影响已经有很多文献讨论,比如Hudson等人1对此作了详细的讨论。图1以Iwaoka等人2研究的羰基氧为例,展示了几种常见的硫-氧孤对电子相互作用模式。
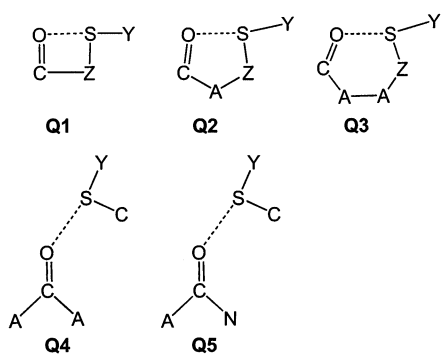
图1. 几种常见的硫-氧孤对电子相互作用模式。大写A表示包括氢在内的任何原子;Y和Z表示C或S(不包括Y和Z同时表示S的情况)。
Hudson等人1用自然键轨道分析方法归属了孤对电子之间不利的相互作用以及硫-孤对电子有利相互作用的成分与来源。理论研究表明,硫原子的孤对电子(lone pair,lp)与氧或氮原子的孤对电子具有排斥的相互作用,然而有时候硫与氮/氧孤对电子之间的有利相互作用可对此补偿并获得净有利的相互作用。这种有利的相互作用来源包括有利的静电(偶极/偶极,dipole/dipole)相互作用以及氧/氮孤对电子与S-Y的σ反键轨道之间的供体-受体轨道相互作用\(X_{lp}\leftrightarrow σ^*_{S-Y}\),其中X是指氧或氮。以图2所示的PDB 4MLH配体VO2为例,在M06-2X/6-31+G(d,p)理论水平用NBO(Natural bond orbital)对母核结构上的硫-氮孤对电子进行计算,量化受体-供体轨道相互作用。在这个例子中,有利的受体-供体轨道相互作用平衡了不利的lp/lp排斥相互作用,并获得净有利的相互作用。与卤键(Halogen bonding)相类似,人们创造了硫键(chalcogen bonding)这个词来称呼硫或其它硫族元素与孤对电子之间这种有利的相互作用,并将之归属为σ-孔相互作用(σ-hole interactions)的一个分支3。
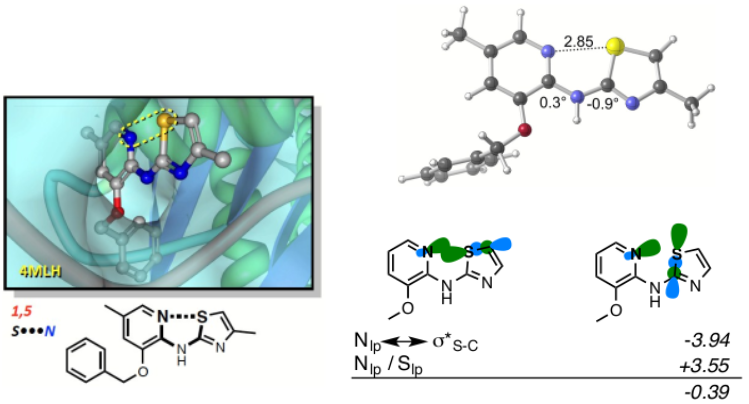
图2. PDB 4MLH配体VO2的硫-孤对电子相互作用1。左下:展示了自然键轨道相互作用及其在硫-孤对电子相互作用中起到的作用。
Hudson等人1认为在对构象和结合模式进行定量或定性预测时忽略硫-孤对电子相互作用是不明智的,即使这些相互作用在能量上不是净有利的。并建议在使用没有考虑硫-孤对相互作用的分子对接方法与打分函数时需要用可靠的量子化学方法进行构象分析、计算构象能。
Bayer公司的Roehrig等人3报道了系列Factor Xa抑制剂,其中17、19(见图1)在化学结构上仅差异一个S/O原子,但活性差异65倍。本文的目的是,以这对分子为例来说明如何用Flare QM的扭转角分析来计算构象能,遵循Hudson等人2建议,用来解释分子内硫-孤对电子相互作用对活性的影响。
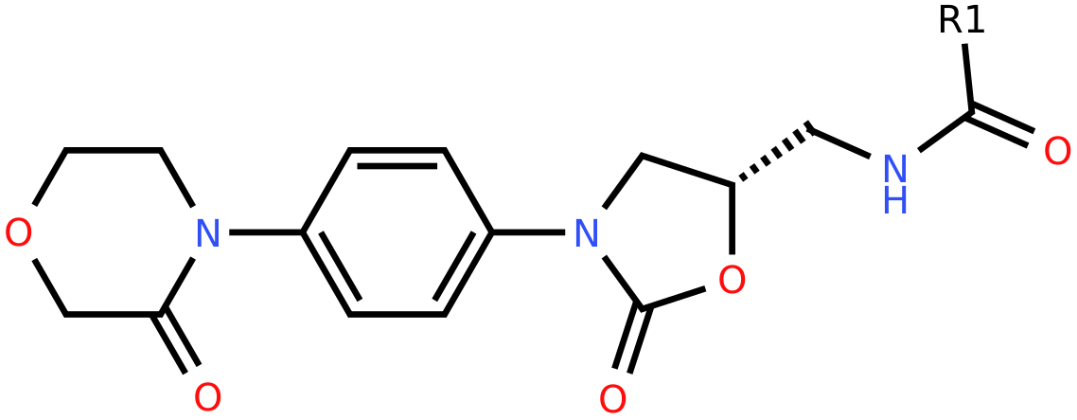
| Compound | R1 | IC50 (nM) |
|---|---|---|
| 17 |  |
0.4 |
| 19 |  |
26 |
图1. Factor Xa抑制剂17与19的化学结构与体外活性
结果
生物活性构象
Bayer公司的Factor Xa抑制剂Rivaroxaban(BAY 59-7939)作为抗凝血药物于2011年获准上市,从其共晶结构PDB 2W26可以发现:其生物活性构象的噻吩环上的硫与酰胺羰基的氧处于同一侧,两面角O=C-C-S=-13°,见图2。
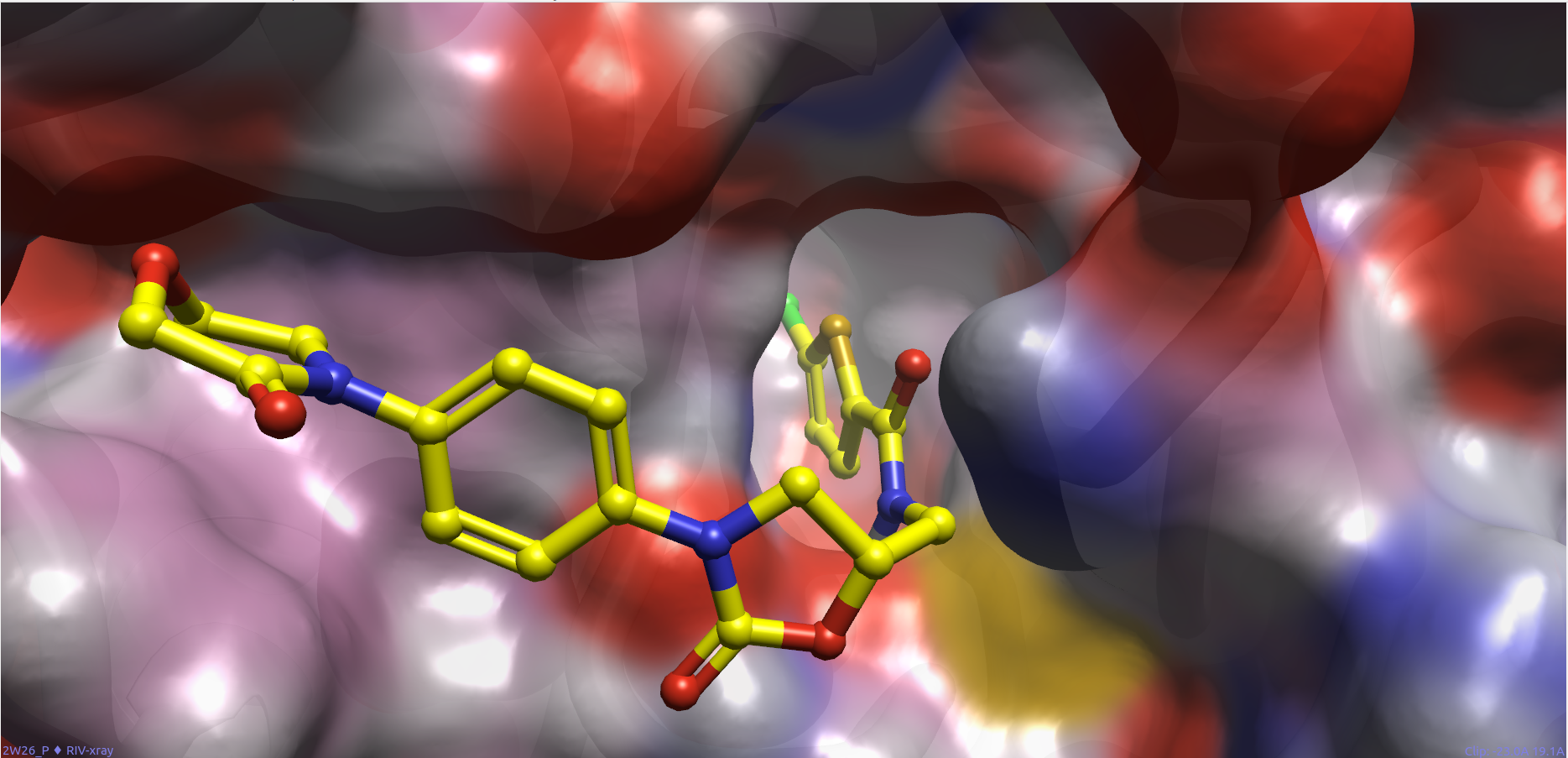
图2. Factor Xa抑制剂Rivaroxaban的共晶结构PDB 2W26。图片为Flare4渲染,其中黄色棍棒:Rivaroxaban;彩色分子表面:Factor Xa。
假设化合物17、19与Factor Xa的结合模式一致,用Flare4在PDB 2W26结合口袋里编辑配体,并用XED力场优化得到化合物17、19的结合模式,结果如图3所示。其中17与19相比,除了噻吩与呋喃的S与O原子坐标不能叠合在一起之外,其它原子的坐标基本一致。
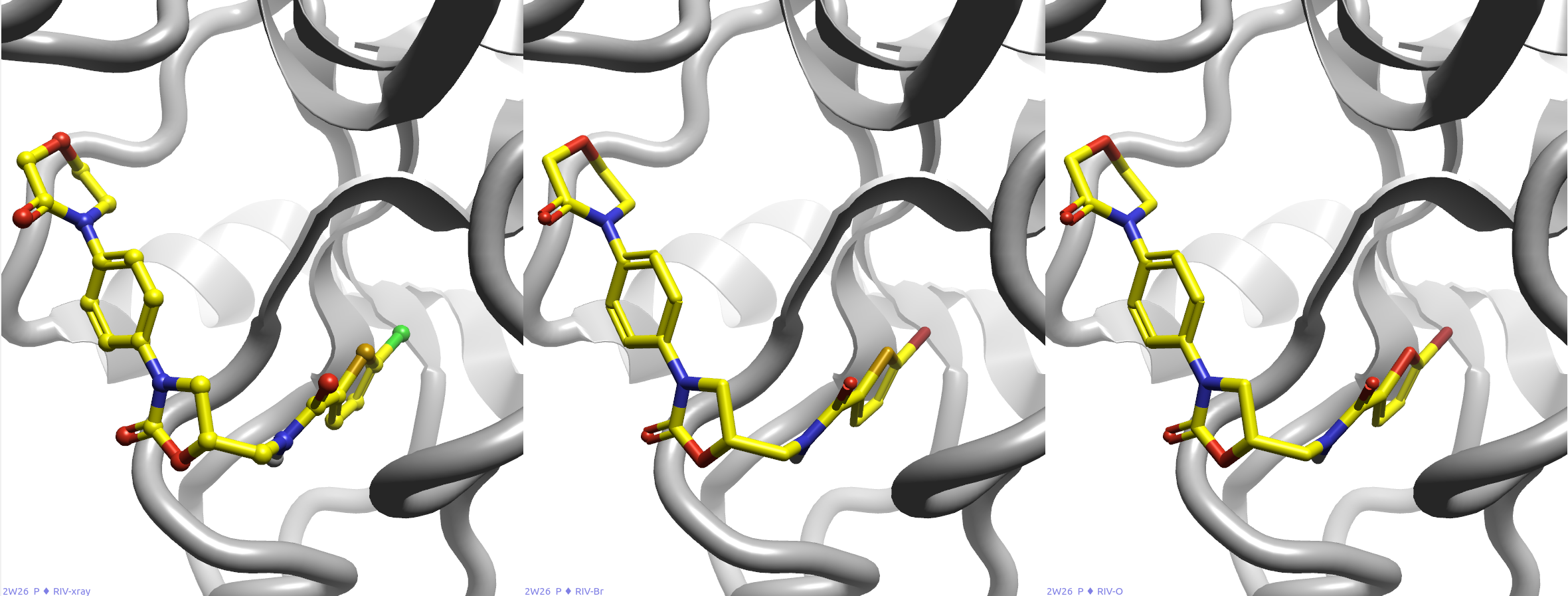
图3. Rivaroxaban(左)、17(中)以及19(右)与Factor Xa的结合模式。灰色飘带:Factor Xa
分子对接打分
GNINA是流行的分子对接软件AutoDock Vina的深度学习打分版本5-7。用GNINA对化合物17、19进行score_only模式的对接计算,计算时将蛋白里面的水全部删除,结果如表1所示。
表1. 化合物17、19的分子对接GNINA打分结果
| Items | 17 | 19 |
|---|---|---|
| gauss 1 | 152.74571 | 154.67422 |
| gauss 2 | 1488.70935 | 1508.62183 |
| repulsion | 8.71463 | 8.34955 |
| hydrophobic | 39.77326 | 40.48775 |
| H-Bond | 1.48635 | 1.33746 |
| Vina score (kcal/mol) | -6.23 | -6.56 |
| CNNaffinity(pKd) | 6.97 | 6.71 |
| CNNscore | 0.98 | 0.96 |
| Factir Xa IC50 (nM) | 0.4 | 26 |
注:AutoDock Vina的打分项为加权之前的值。
17与19相比,它们具有相似大小的相互作用项,因此它们具有相似的Vina score打分值,预测的结合自由能分别为-6.23与-6.56kcal/mol,没有显著的差异。相似的,GNINA深度学习打分CNNaffinity对17、19的结合亲和力预测值(pKd)分别为6.97与6.71,也没有显著差异。这说明,无论是经典的Vina打分、还是深度学习亲和力打分都不能解释实验观察到的IC50差异。
基于QM的扭转角分析
并不直接对化合物17、19进行基于QM的扭转角分析,而是用图4简化的模型分子代替原型化合物进行计算。先用Flare QM在BP86-D3BJ/DEF2-TZVP理论水平下对模型分子进行几何优化,然后在同样的理论水平下进行扭转角分析,每隔5°进行扫描,结果如图4所示。在生物活性构象时(两面角~-15°),17与19的扭转角张力能分别为0.17与3.48kcal/mol,17比19的扭转角张力能低了约3.3kcal/mol。在与Factor Xa以同一个模式结合时,17受到的张力能惩罚比19的低3.3 kcal/mol,这可用来解释为什么17的体外活性比19的高。
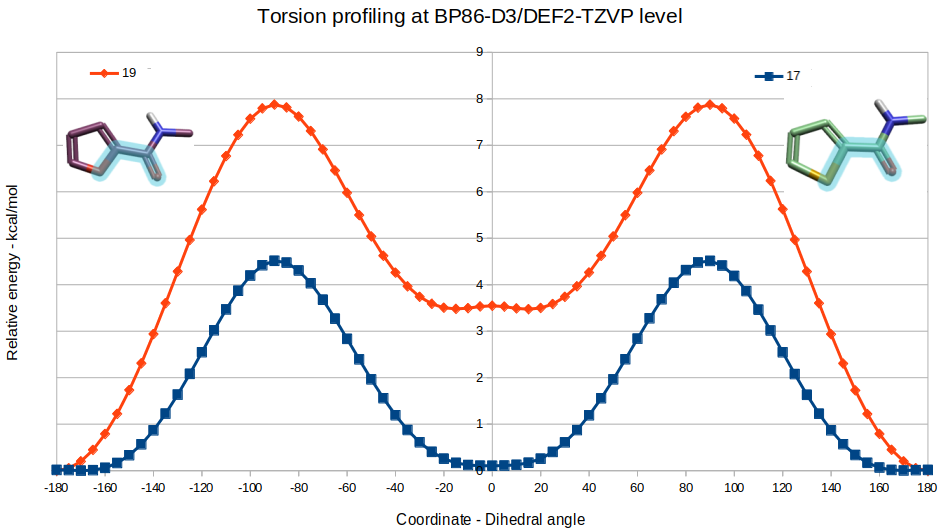
图4. 化合物17、19的模型化合物及其在BP86-D3BJ/DEF2-TZVP理论水平下的扭转角分析。高亮的原子:在扭转角分析时用来定义两面角的四个原子。
方法
蛋白结构与配体结构的准备
将Rivaroxaban与Factor Xa的共晶结构PDB 2W26下载到Flare V8,并用Protein Prep工具进行结构准备以添加氢原子、优化氢键、消除原子冲突并给蛋白结构与配体分配最佳质子化状态,并对截短的蛋白质链继进行封端。从准备好的复合物结构里将配体提取出来,得到Rivaroxaban的生物活性构象与蛋白结构。在Flare里复制配体Rivaroxaban,原子替换的方式继续编辑,得到化合物17、19,然后在蛋白背景下用XED力场对化合物17、19并进行精确模式的能量最小化。
模型分子的准备
模型分子是从上一步准备好的活性构象配体基础上截取得到,截小后的模型分子保留其构象与生物活性构象一致,并作为后续Torsion Profile分析的起点。
QM几何优化
QM几何优化使用Flare QM的Minimize在BP86-D3BJ/DEF2-TZVP理论水平下计算。
QM扭转角分析(Torsion Profile)
用Flare QM的Torsion Profile在BP86-D3BJ/DEF2-TZVP理论水平下对模型分子的两面角进行扭转角分析,计算参数如下:
- Method: DFT
- DFT functional: BP86
- Use dispersion correction:Yes
- Basis set: def2-tzvp
- Convergence: Medium
- Max iterations: 500
- Number of rotomers: 72
- Degrees: 5.0°
- Max Threads: Auto
其中Degrees是根据Number of rotomers自动生成,无需主动设置,这里意味着以5.0°为步长进行两面角扫描。
结论
本文以一对结构仅一个原子差异但活性有65倍差异的Factor Xa抑制剂为例,分别用GNINA分子对接与Flare QM扭转角分析对结合模式进行打分与构象能计算,结果表明:不考虑分子内硫-孤对电子相互作用的分子对接打分函数不能区分这对分子的实验活性差异;基于QM的扭转角分析可以捕捉到有利的硫-孤对电子相互作用,发现扭转角张力能差异,并用来解释实验观察到的活性差异。分子内硫-孤对电子相互作用对于小分子药物设计以及与受体的相互作用影响不可忽略,Flare QM可以方便地进行此类相互作用研究。
文献
- Hudson, B. M.; Nguyen, E.; Tantillo, D. J. The influence of intramolecular sulfur-lone pair interactions on small-molecule drug design and receptor binding. Org. Biomol. Chem. 2016, 14, 3975−80.
- Iwaoka, M., Takemoto, S. and Tomoda, S. Statistical and Theoretical Investigations on the Directionality of Nonbonded S···O Interactions. Implications for Molecular Design and Protein Engineering. Journal of the American Chemical Society. 2002, 124(35),10613–10620. https://doi.org/10.1021/ja026472q.
- Politzer, P.; Murray, J. S.; Clark, T. Halogen bonding and other σ-hole interactions: a perspective. Physical Chemistry Chemical Physics. 2013, 15(27), 11178. https://doi.org/10.1039/c3cp00054k
- Roehrig, S. et al. Discovery of the Novel Antithrombotic Agent 5-Chloro-N-({(5S)-2-oxo-3- [4-(3-oxomorpholin-4-yl)phenyl]-1,3-oxazolidin-5-yl}methyl)thiophene- 2-carboxamide (BAY 59-7939): An Oral, Direct Factor Xa Inhibitor. Journal of Medicinal Chemistry. 2005, 48(19), 5900–5908. https://doi.org/10.1021/jm050101d.
- Flare V8. https://www.cresset-group.com/software/flare
- Trott, O. and Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of Computational Chemistry, 2009, 31(2), 455–461. Available at: https://doi.org/10.1002/jcc.21334.
- Sunseri, J., & Koes, D. R. Virtual Screening with Gnina 1.0. Molecules, 2021, 26(23), 7369. https://doi.org/10.3390/molecules26237369
- GNINA 1.0, https://github.com/gnina


