
选择性CDK7抑制剂SY-5609的大环化设计
摘要:Flare,由Cresset公司研发的综合性药物设计软件,在最新版本V9中集成了Spark模块,后者是一个基于片段的分子设计工具。本文评估了Flare/Spark在大环化合物设计领域,尤其是针对CDK7抑制剂SY-5609的大环化设计中的表现。结果表明,SY-5609中嘧啶-吲哚片段的生物活性构象与量子力学计算预测的最低能量状态具有高度一致性,且90°的二面角实际上对应于局部能量的最大值,而非先前文献中所报道的最小值,这一发现得到了单晶X-衍射数据的验证。此外,Flare有效地阐释了SY-5609对CDK7相较于CDK2的高选择性机制,并准确预测了大环化改造可能对这种选择性产生的影响,该预测结果与后续的实验数据一致。利用Spark进行的大环设计实验不仅成功复现了已知的连接臂化学类型,还揭示了新的连接臂结构,这些新结构均保留了SY-5609与CDK7关键残基间的相互作用。相较于人工智能生成的设计,Spark的设计更为紧凑,更符合预设的设计标准,从而证明了Flare/Spark作为一个高效的大环化合物设计平台,对于需要精确控制分子构象以优化生物活性和选择性的药物设计项目具有显著的应用价值。
肖高铿/2024-11-16
前言
大环化合物具有多种优势,比如对刚性靶标具有高亲和力和选择性,以及改善药物类性等特点。最近,Lu等人1报道了利用其AI驱动的药物设计平台Chemistry24,从选择性CDK7抑制剂SY-5609(化合物1,图1)出发,通过大环化策略开发新型的非共价CDK7抑制剂的方法,高效地发现了具有新颖大环骨架的CDK7抑制剂2与3(表1)。经过对3进行结构-活性关系(SAR)研究和ADME优化后,获得了化合物13(图1),该化合物表现出强效的体外活性、良好的ADME性质以及在异种移植瘤模型中显著的体内抗肿瘤活性。
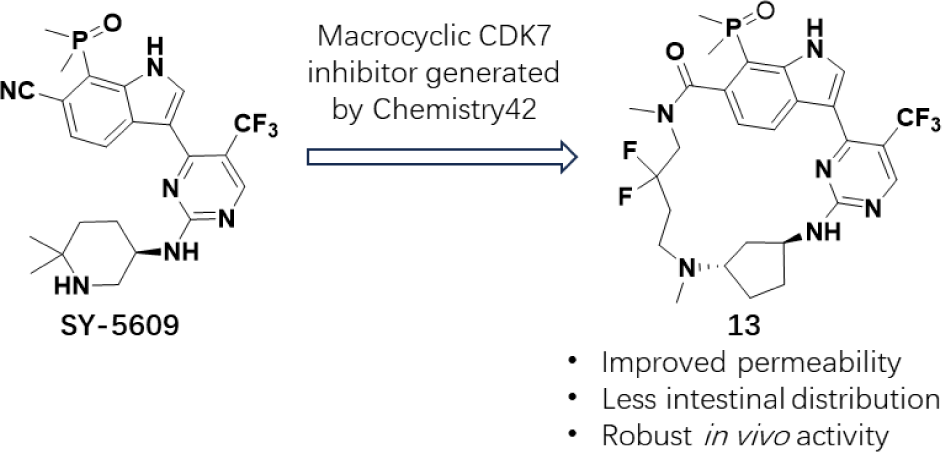
图1. SY-5609(化合物1)及其大环化衍生物13,图片来源于文献[1]
Lu等人1的大环化设计源于对SY-5609与CDK7的结合模式假设:SY-5609与CDK7结合时其嘧啶-吲哚两面角为32.4°(图2-A),分子动力学模拟表明该两面角稳定在~40°,而其最低能构象的两面角为90°,因此构建大环化合物使得生物活性构象与最低能构象一致将有利于活性的提升。
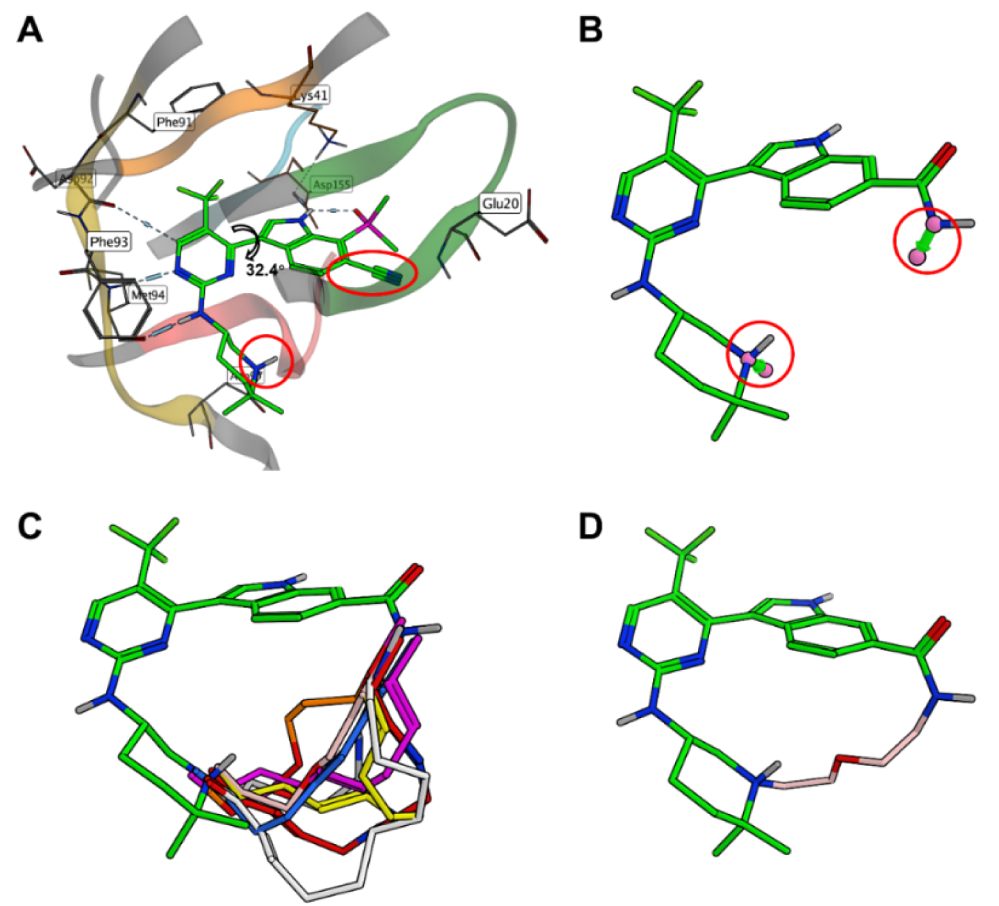
图2. SY-5609的大环化设计,图片来源于文献[1]
在Lu等人1的第一轮设计中,将SY-5609的腈基修改为酰胺,然后以酰胺N与哌啶N为大环连接臂的连接点(图2-B),生成320个不同的连接臂(图2-C),最终选择连接臂最短的那个(图2-D)为研究起点。
表1. 苗头化合物2与3及其生物学活性。
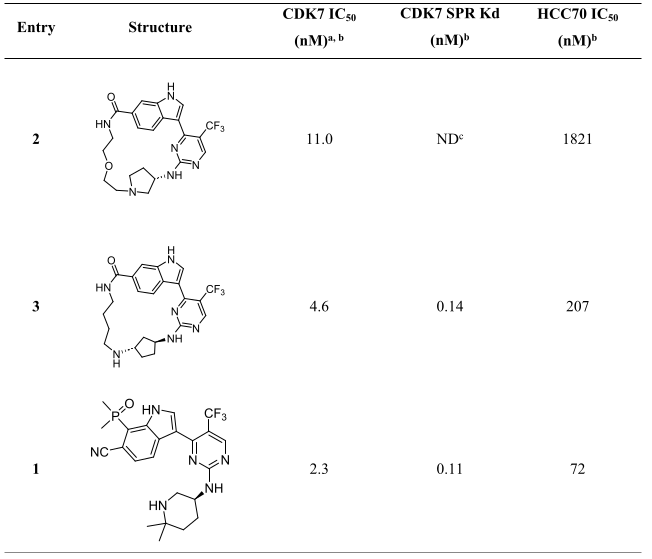
在Lu等人1的第二轮设计中以图2-D为起点,在保留连接点的前提下,增加了碱性原子中心与疏水中心特征。在这一轮设计中获得了化合物2与3(见表1)。由于化合物2、3结合构象(模拟)的嘧啶-吲哚两面角τ分别与它们全局最低能构象的两面角τ接近,因此认为这两个化合物有潜力保留甚至改善对CDK7的活性。生物活性测试结果如表1所示,其中化合物2在HCC70细胞系上的活性表现不佳(IC50=1821nM),而化合物3表现出良好的酶学、细胞水平活性。作者进一步以3为苗头开展SAR研究,最终发现了活性与成药性均得以改善的化合物13(见图1)。
本文的主要目的是以SY-5609生物活性构象为起点分子,尝试使用药物设计软件Flare/Spark的大环化设计工作流,考察其能否设计出合理的大环苗头化合物,并用Lu等人1的研究结果进行回溯性地验证。
结果
生物活性构象分析
将SY-5609及其类似物与CDK7/CDK2的共晶结构(PDB:8R9U、7RA5、6XD3)下载到Flare,进行标准的蛋白结构准备,然后分析SY-5609嘧啶-吲哚生物活性构象的两面角(τ),可以发现它们分布在-25~-35°之间,如图3所示。
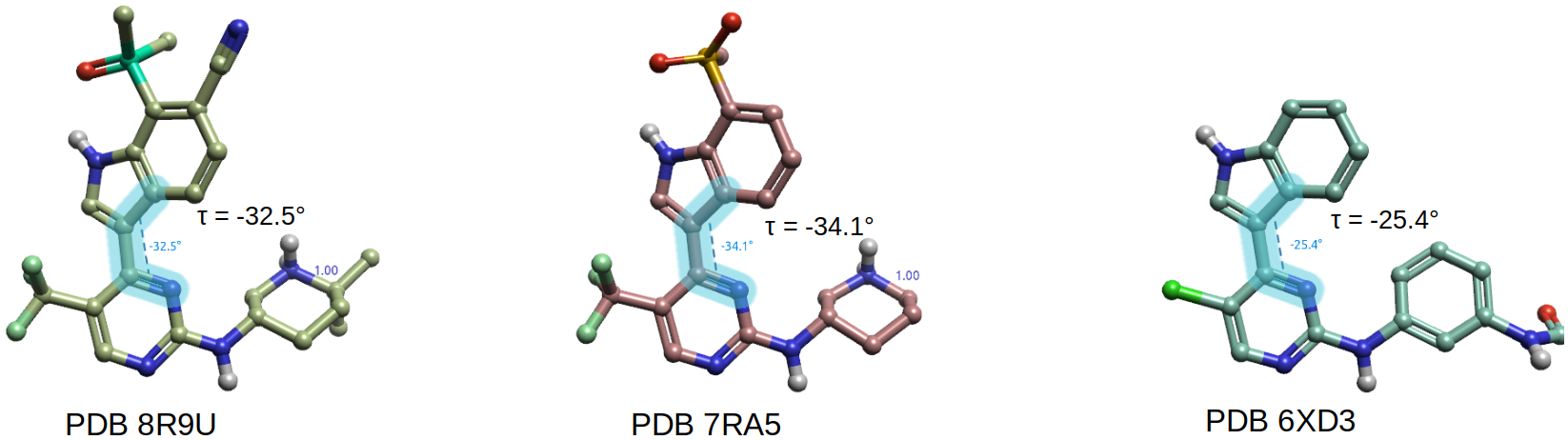
图3. SY-5609嘧啶-吲哚生物活性构象的两面角(τ)分布范围
表2给出了共晶结构配体的生物活性构象嘧啶-吲哚两面角(τ)与CDK7以及CDK2激酶活性的关系。可以发现,非共价抑制剂与CDK7(PDB 8R9U)、CDK2(PDB 7RA5)相结合时,嘧啶-吲哚片段具有几乎一样的两面角,τ ~ -33°。化合物SY-1356(PDB 6XD3)以共价方式与CDK7结合,两面角τ = -25°。
表2. SY-5609的嘧啶-吲哚两面角(τ)在生物活性构象中的分布总结
| PDB ID | Target | τ | ΔE(kcal/mol) | CDK7 SPR Kd(nM) | CDK2 IC50(nM,b) |
|---|---|---|---|---|---|
| 8R9U | CDK7 | -32.5° | 0.31 | 0.072 | 55242 |
| 7RA5 | CDK2 | -34.1° | 0.43 | 0.072 | 782 |
| 6XD3 | CDK7 | -25.4° | 0.06 | 17.43,a | \(>2000\)3 |
Target:共晶结构的蛋白;a:Ki;b:[ATP] = 2 mM;ΔE:构象张力能
SY-5609嘧啶-吲哚片段的扭转角分析
用Flare内置的量子力学计算方法在BP86-D3BJ/DEF2-TZVP理论水平对图3模型分子嘧啶-吲哚四个高亮原子定义的两面角进行扭转角分析,结果如图3所示:在τ=-20°~-90°区间,随着角度的增加(越接近直角),张力能单调上升。当τ=-20°时,张力能最低(ΔE = 0.0 kcal/mol),当τ=-90°时,张力能最高(ΔE = 5.0 kcal/mol)。生物活性构象的两面角为高亮显示的绿色区域,其张力能为0.08~0.43 kcal/mol。代表三个共晶结构(PDB:8R9U、7RA5、6XD3)配体的模型分子三氟甲基嘧啶-吲哚的张力能分别为0.31、0.43、0.06 kcal/mol(见表2),这是一个相对低的值,因此可以认为嘧啶-吲哚两面角的生物活性构象是相对稳定的。
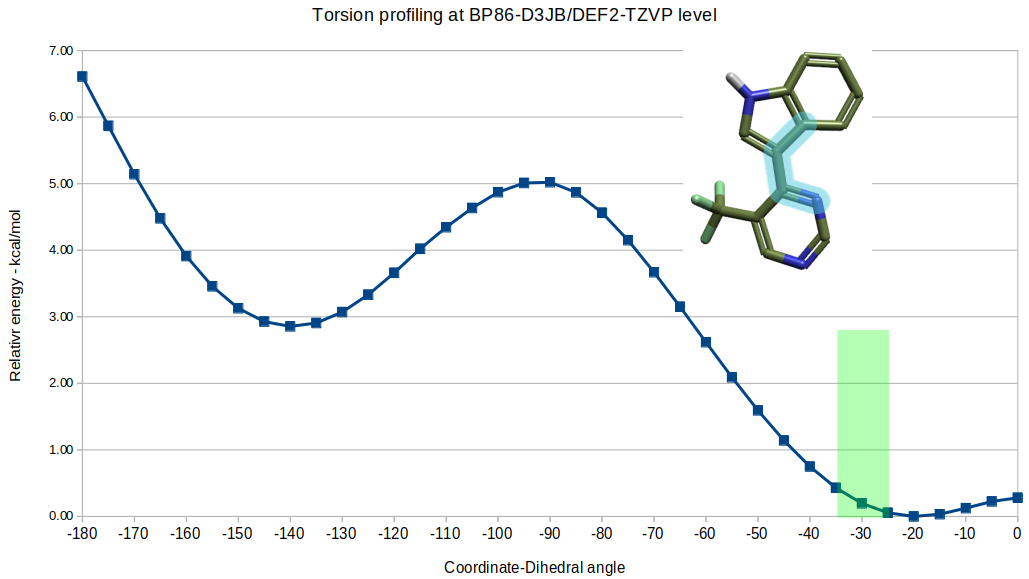
图3. SY-5609的嘧啶-吲哚两面角基于QM的扭转角分析。绿色区域:生物活性构象的两面角分布;棍棒模型:扭转角分析用的模型分子。
虽然图3基于QM的扭转角分析仅显示了τ在0~-180°范围内的相对能量,但是模型分子τ在0~180°范围内构象的相对能量可通过对称操作来推断。基于QM扭转角分析的结果可得到Marineau等人2公开的SY-5609晶体结构数据(CCDC:2093192)的支持。图4显示了SY-5609单晶X-衍射发现的四个构象(CCDC 2083192)及其嘧啶-吲哚两面角τ的值,其中右上与右下构象的两面角τ~+/-20°,与QM计算的最低能两面角一致;其中左上与左下构象的两面角τ~+/-30°,相对能量不到0.2 kcal/mol。
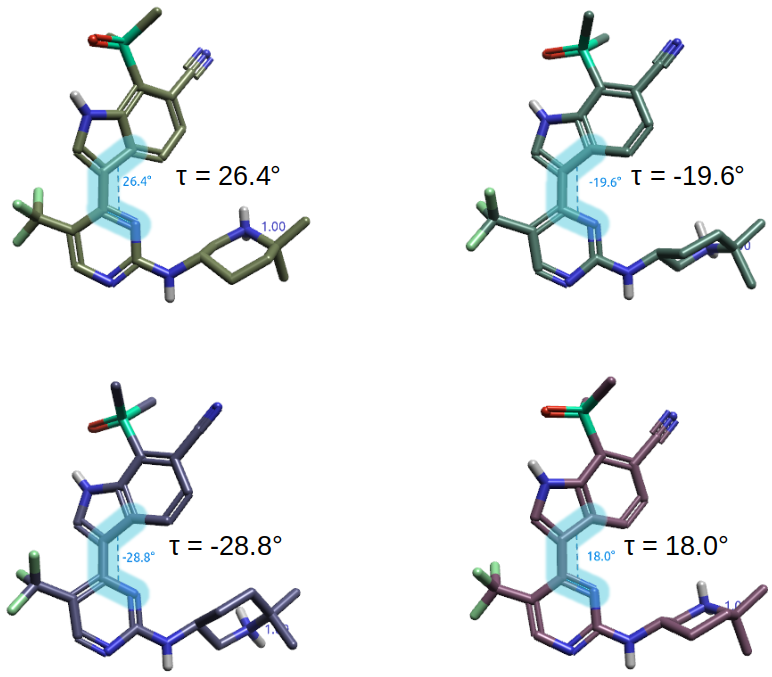
图4. SY-5609单晶X-衍射发现四个构象(CCDC 2083192)及其嘧啶-吲哚两面角τ
如前所述,当嘧啶-吲哚两面角τ = -90°时,是局部最高能的构象(ΔE = 5 kcal/mol),这与Lu等人1认为当τ = 90 °是最低能构象的结论相反。总的来说,SY-5609的单晶衍射数据与嘧啶-吲哚片段的QM扭转角分析结果均不支持Lu等人1提出的嘧啶-吲哚两面角的生物活性构象是高能构象的假设。
SY-5609靶标选择性分析
在Flare里采用基于配体的方法4将SY-5609类似物与CDK2共晶结构PDB 7RA5叠合到SY-5609与CDK7的共晶结构PDB 8R9U上,结果如下图4所示。可以看到公共骨架部分结合模式几乎完全一样,尤其是关键的极性相互作用,比如化合物与激酶铰链区的氢键结合模式,哌啶环正电中心氮与Asp末端羧基的盐桥相互作用,吲哚-NH与Asp末端的氢键相互作用等等。
图4.左边:SY-5609/CDK7共晶结构PDB 8R9U;右边:类似物/CDK2共晶结构PDB 7RA5。使用:将鼠标悬停在图片上,点击播放按钮。
比较PDB 8R9U(CDK7)与7RA5(CDK2)共晶配体,除了吲哚环上的取代基之外,公共部分的原子几乎重和在一起,如图5所示。如表2所示,两个配体对CDK7具有同样的亲和力(具有同样的Kd值,见表2)而对CDK2具有截然不同的生物活性,这个靶标选择性可以归因于它们在吲哚苯环上的取代模式差异。
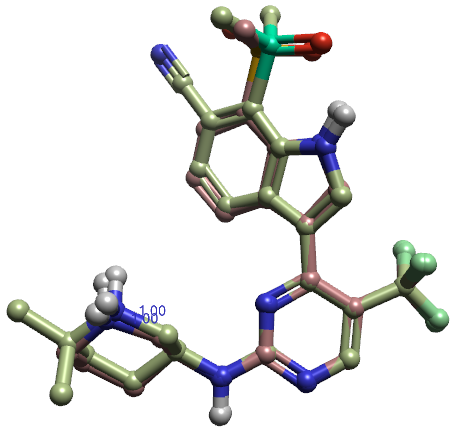
图5. PDB 8R9U(CDK7)与7RA5(CDK2)共晶配体的比较。
采用基于配体的方法4将CDK2共晶结构PDB 7RA5叠合到CDK7的共晶结构PDB 8R9U上,然后比较SY-5609(8R9U的共晶配体)与两个靶标的相互作用模式。如图6所示,SY-5609与CDK7、CDK2相互作用最大的差异在于吲哚环上腈基与CDK2的GLY11、GLU12(图6-右高亮氨基酸残基)发生了严重的空间立体冲突,这可用来解释SY-5609对CDK7/CDK2选择性(IC50分别为0.18、5524nM)。
图6. CDK7(PDB 8R9U)与7RA5(CDK2)结合口袋的比较。左:飘带为CDK7,球棍为SY-5609;右:飘带为CDK2,球棍为SY-5609,高亮残基为GLY11与GLU12。
评估大环化合物的靶标选择性
在Lu等人1的研究中,提供了大环化合物2、3(表1)与CDK7结合的计算模型,将这两个模型按Cα叠合到PDB 8R9U,并与同样叠合到PDB 8R9U的CDK2(PDB 7RA5)结合口袋进行比较,结果如图7所示:可以发现大环化合物2、3的吲哚环部分并没有与CDK2的GLY11、GLU12发生立体冲突。这意味着大环化合物2、3可能对CDK2具有良好的抑制活性。尽管Lu等人1的研究没有提到大环合物2、3的CDK2活性,但是可从3的衍生物13来了解到。大环化合物13对CDK7与CDK2的IC50分别为0.18、30.3nM。这说明大环化合物确实降低了对CDK2的选择性,对CDK2的IC50从SY-5609的5529nM降低到化合物13的30.3nM。这也进一步证明了前面关于靶标选择性的假设是正确的。
图7. 大环化合物2、3与CDK2(PDB 7RA5)的结合模式。飘带:CDK2(PDB 7RA5),高亮残基为GLY11与GLU12;球棍模型(左):化合物2;球棍模型(右):化合物3。
需要注意的是:文章提供化合物13对CDK2、CDK7的IC50分别为30、0.18 nM,在Figure 4的表格A里提供了CDK7/CDK2的选择比为168倍。但是从文章提供的实验部分并不知道关键的实验条件[ATP],我猜测在两个体外实验中[ATP]是在各自Km,而这不能反映真正靶标的选择性。
用Spark进行大环化设计
将PDB 8R9U下载到Flare,进行标准的蛋白结构准备,并在结合位点里对共晶配体(SY-5609)的腈基进行编辑,进行几何优化,得到图2-B的起点分子。然后在 Flare 里使用 Spark 的Macrocyclization Wizard开始大环化设计。选中酰胺N上朝向哌啶环的那个氢、并选中哌啶环,如图8所示,被选中的部分作为大环化设计中需要被连接臂替换掉的部分。
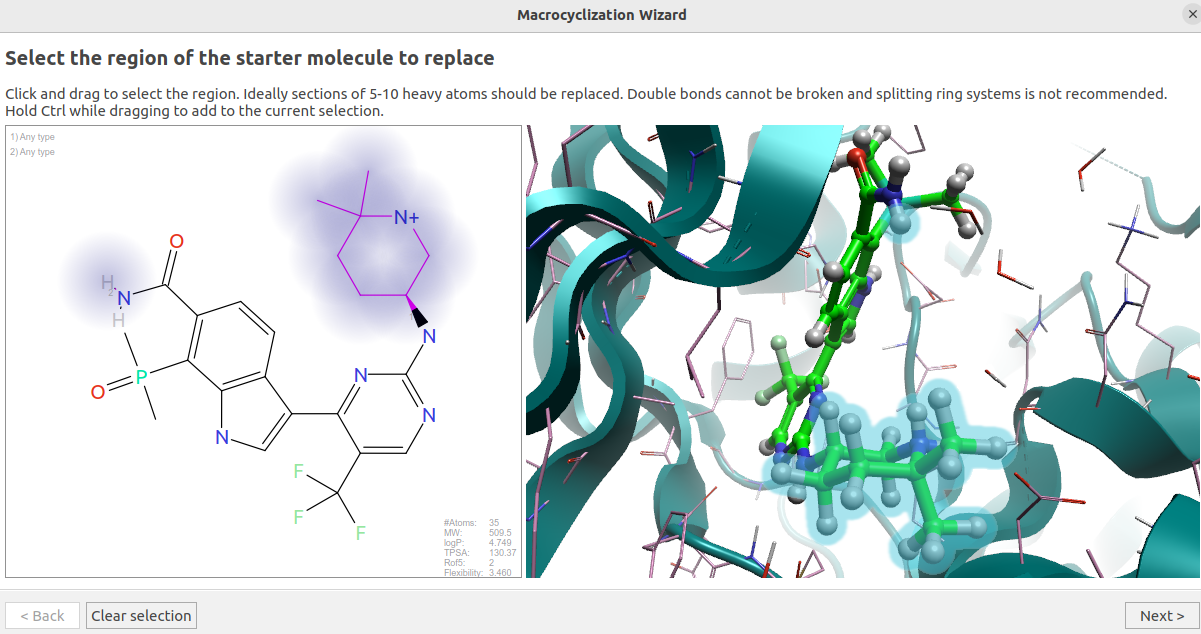
图8. Spark 大环化设计的起点分子以及连接点。左边2D视窗:高亮部分是要大环设计中要被替换的部分,连接点1是与嘧啶环2位环外氮原子连接那个哌啶环碳原子,连接点2是酰胺氮上指向哌啶环的氢原子。右边3D视窗:实时观察被选中的部分与连接点,以确保连接点的选择是正确的。
为了尽可能让 Spark 大环设计与Lu等人1报道的策略一致,对连接点与选项进行了如下设置:
- 连接点1:Csp3,ring only
- 连接点2:Csp3,acyclic only
- filters/contains a ring:require
- filters/contains an aromatic ring:exclude
与嘧啶2位的环外氮原子连接的连接臂原子必须是sp3杂化的碳原子,并且是一个环上的原子。
与酰胺氮原子连接的连接臂原子必须是sp3杂化的碳原子,并且不能是环上的原子。
连接臂至少含有一个环。
连接臂不得含有芳香环。
在本次大环设计的计算实验中,选择了38个片段数据库,共包含218万个化合物片段(图9),大环连接臂将从这些片段中产生。计算采用基于配体的打分函数,其它参数均为软件预设参数,默认保留打分最高的500个分子。
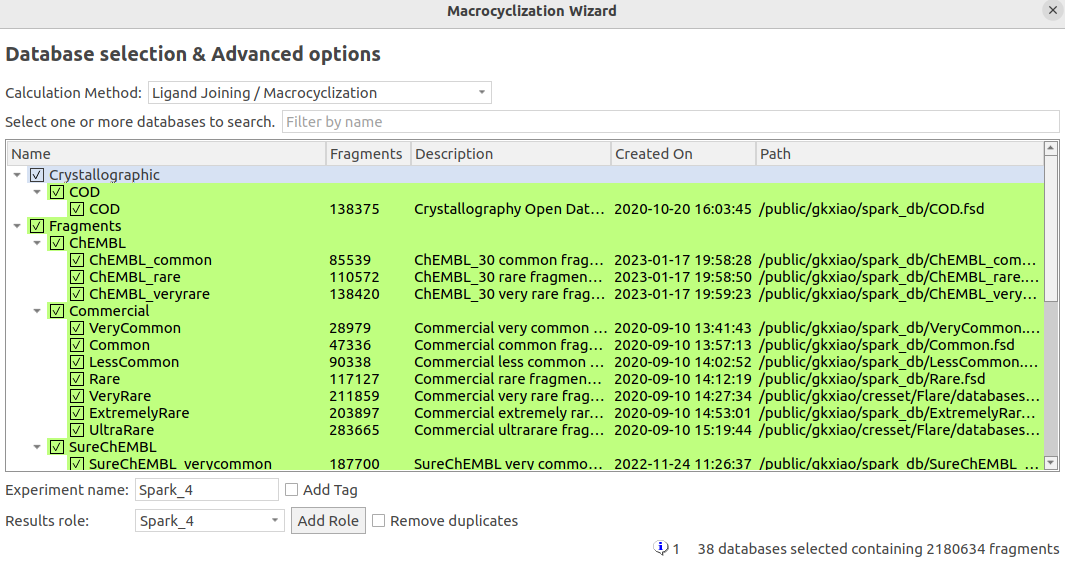
图9. Spark大环化设计的片段数据库。
Spark大化实验总共生成了500个结构,XED场相似打分值在0.784-0.853之间。其中31个连接臂含有与化合物2类似四氢吡咯子结构(以下简称2-类似物),13个连接臂含有与化合物3类似的氨基环戊烷子结构(以下简称3-类似物)。
图10展示了部分2-类似物,其中一个显著特点是连接臂中的四氢吡咯氮原子作为正电中心,模拟了SY-5609中哌啶环上带正电的氮原子,并与CDK7残基Asp97末端的羧基形成了盐桥相互作用。另一个特点是,连接四氢吡咯氮原子与酰胺氮原子的链长范围为3至4个原子,这与Lu等人1所报道的5个原子长度有所不同。
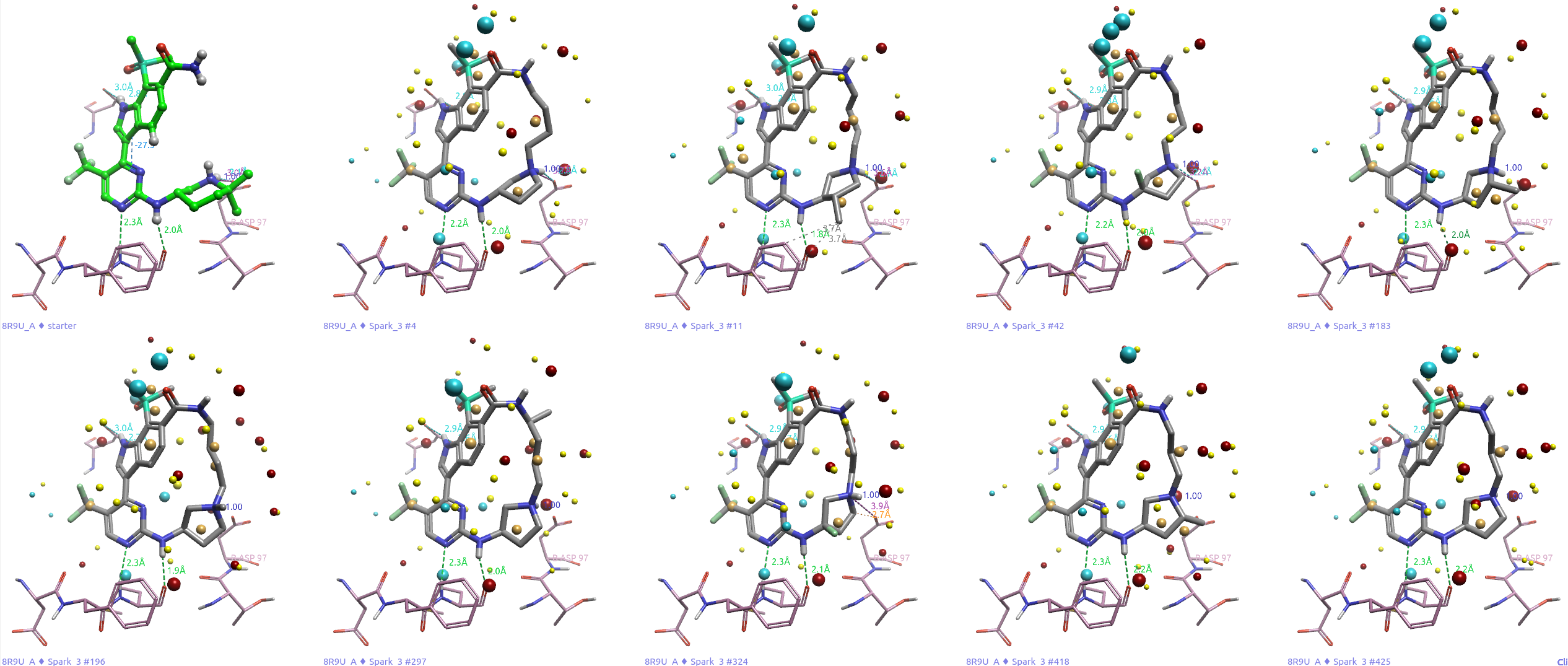
图10. 含四氢吡咯子结构的连接臂示例。
图11展示了部分3-类似物,其显著特征之一为连接臂中包含一个带正电荷的氨基环戊烷,该结构通过环外的正电荷模拟了SY-5609中哌啶环上带正电荷的氮原子,并与CDK7残基Asp97末端的羧基形成盐桥相互作用。另一特点是,连接氨基环戊烷氮原子与酰胺氮原子的链长介于2至3个原子之间,相较于Lu等人1报告的4至5个原子长度要短。此外,1,3-取代的环戊烷存在顺式和反式异构体的差异,这种差异进一步影响了其与Asp97末端羧基形成盐桥相互作用的可能性。
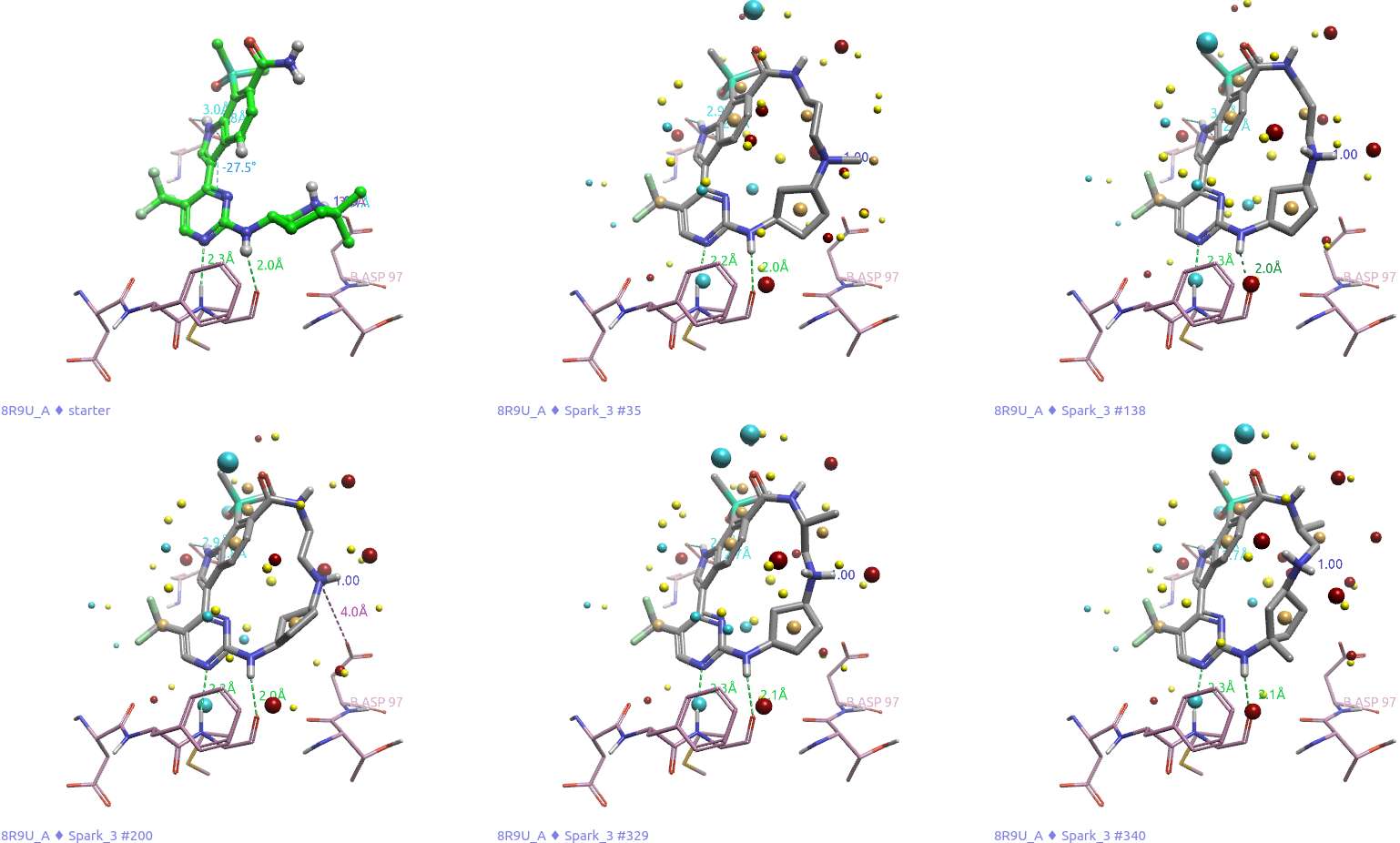
图11. 含氨基环戊烷子结构的连接臂示例。
在Spark的结果中,自然也包括了与起始化合物相同的质子化哌啶环连接臂。图12展示了几种此类连接臂的实例,其中哌啶氮原子与酰胺氮原子之间的链接长度根据哌啶的取代模式及构象不同而有所变化,通常在3至4个原子范围内。值得注意的是,这些连接臂均与CDK7残基Asp97末端的羧基形成了盐桥相互作用。
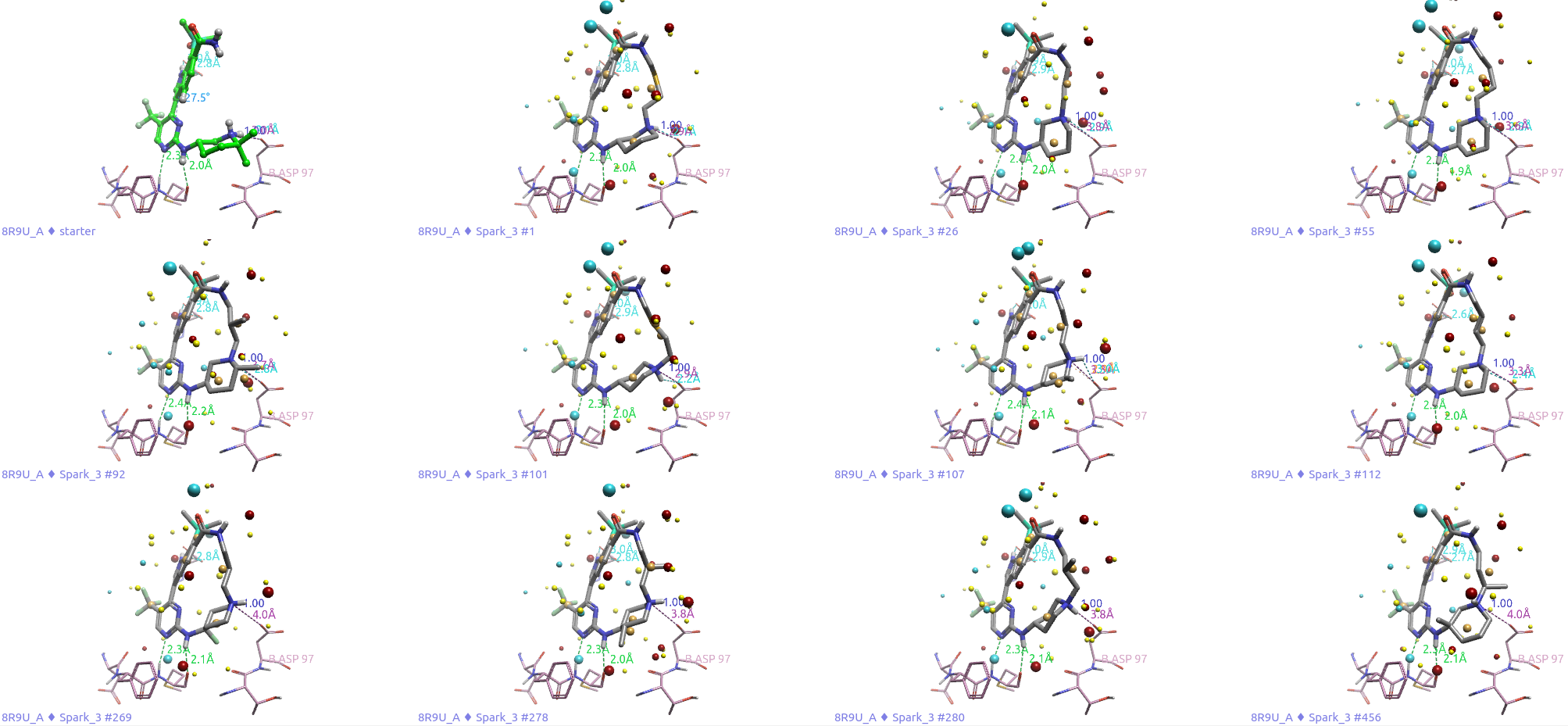
图12. 含质子化哌啶环的连接臂示例。
除了上述已知的化学类型外,Spark的结果中还包含许多新颖的连接臂。图13展示了一些新型连接臂的例子,如四元环和复杂的螺环结构,这些结构均与CDK7残基Asp97的末端羧基形成了盐桥相互作用。
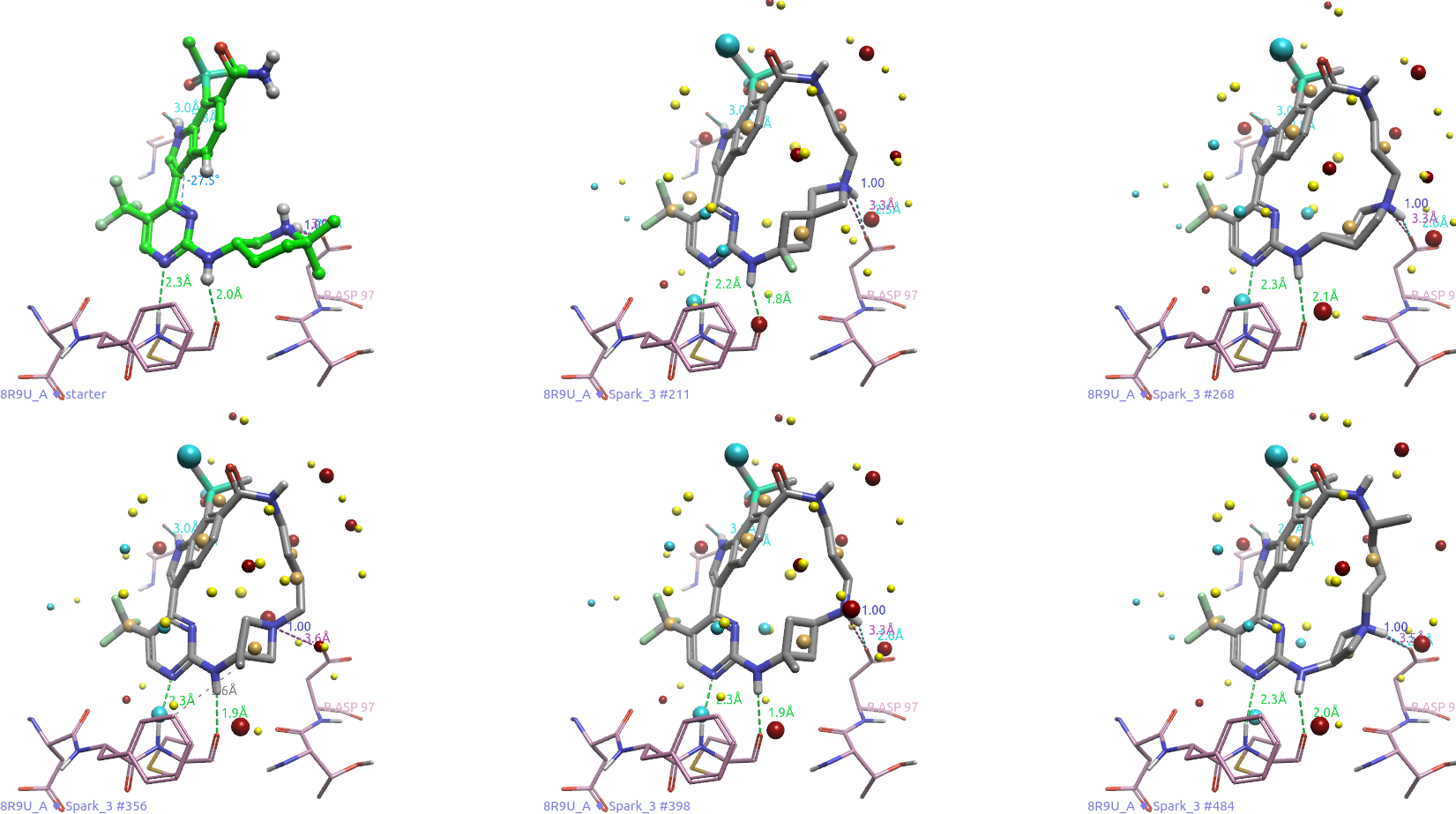
图13. 部分新型的连接臂示例。
此外,Spark的大环设计方法比AI方法表现出更高效的性能。Lu等人1以更短的连接臂为目标挑选潜在的连接臂,经过两轮的AI生成设计才锁定苗头化合物2与3。而Spark仅需一轮,就得到连接臂更短的苗头化合物。
总体而言,在此次大环化计算实验中,Spark不仅重现了Lu等人1报道的化学结构类型,还给出了多种未见文献报道的新颖化学结构。这些新结构同样能够模拟起始化合物SY-5609与CDK7之间的关键相互作用,为后续筛选工作提供了宝贵的创意来源。然而,在采纳某一创意之前,建议先行评估其构象稳定性,以确保大环化合物能够稳定保持生物活性构象,进而判断是否有必要投入较高的合成成本进行化合物的制备。鉴于大环化合物的合成难度和成本通常高于线型化合物,此类评估尤为重要。有关如何进行构象稳定性评估的详细讨论,可在本站点搜索“conformational design”获取更多信息。
结论
Flare 是 Cresset 公司开发的一款融合了基于结构与基于配体的药物设计功能的软件,最新发布的版本 V9 集成了 Spark 基于片段的分子设计工具。本文旨在利用 Lu 等人1关于 CDK7 抑制剂 SY-5609 的大环化设计数据,回顾性地评估 Flare V9 在大环化合物设计方面的性能表现。
首先,本文用 Flare 对结构生物学数据进行了分析,统计了 SY-5609 中嘧啶-吲哚片段的生物活性构象二面角分布范围。随后,对同一片段进行了基于QM的扭转角分析。结果显示,生物活性构象与量子力学计算得出的最低能量构象高度一致,且与 SY-5609 单晶衍射数据相一致。值得注意的是,90° 二面角实际上对应于局部能量最大值,这与 Lu 等人1提出的90°二面角是最低能构象假设相悖。
基于配体的蛋白质叠合分析显示,Flare 能够有效地解析 SY-5609 对 CDK7 相对于 CDK2 的高选择性机制,并准确预测大环化改造可能降低对 CDK2 的选择性,这一结论得到了 Lu 等人1生物活性数据的支持。
在使用 Spark 进行的大环设计计算实验中,我们不仅成功复现了文献中报道的连接臂化学类型,还发现了多种新颖的连接臂化学结构。所有这些新发现的结构均保留了 SY-5609 与 CDK7 关键残基之间的相互作用特征。此外,与人工智能生成的大环连接臂相比,在 Spark 的结果中包含有更短的连接臂,更符合预先设定的设计要求,展现了出色的大环设计效率。
综上所述,集成有Spark模块的Flare提供了一套全面的大环化合物设计解决方案,能够高效完成大环设计任务。本研究证明了Flare在探索新药先导化合物方面具备强大的潜力,特别是对于那些需要精确控制分子构象以优化生物活性和选择性的药物设计项目。
附件
附件包含了已经叠合好的蛋白与配体结构。
1 2 3 4 5 6 7 8 9 | cdk7-sy5609-macrocyclization ├── 6xd3.pdb ├── 7b5o_AltaA_ligand.pdb ├── 7b5o_AltaA_prot.pdb ├── 7ra5_ligand.sdf ├── 7ra5_prot.pdb ├── 8r9u_ligand.sdf ├── 8r9u_prot.pdb └── SY-5609-macrocyclization_SI.flr |
下载链接: 附件
文献
- Lu, H. et al. (2024) “Discovery of a Novel Macrocyclic Noncovalent CDK7 Inhibitor for Cancer Therapy,” Journal of Medicinal Chemistry [Preprint]. Available at: https://doi.org/10.1021/acs.jmedchem.4c02098.
- Marineau, J.J. et al. (2022) “Discovery of SY-5609: A Selective, Noncovalent Inhibitor of CDK7,” Journal of Medicinal Chemistry, 65(2), pp. 1458–1480. Available at: https://doi.org/10.1021/acs.jmedchem.1c01171.
- Hu, S. et al. (2019) “Discovery and Characterization of SY-1365, a Selective, Covalent Inhibitor of CDK7,” Cancer Research, 79(13), pp. 3479–3491. Available at: https://doi.org/10.1158/0008-5472.CAN-19-0119.
- 用Flare Python API实现根据配体叠合来调整蛋白的取向. http://blog.molcalx.com.cn/2023/04/23/protein-transformation.html
联系我们
想在自己的项目中使用Spark大环化设计或使用Flare进行数据分析,请联系我们获取免费的试用版。
你还可以采购软件或委托我们进行项目合作:info@molcalx.com。
原创文章,作者:小墨,如若转载,请注明出处:《选择性CDK7抑制剂SY-5609的大环化设计》http://blog.molcalx.com.cn/2024/11/16/cdk7-inhibitor-sy-5609-macrocyclization.html


