
Flare V6发布——新特性、新功能与改进
摘要:本文介绍了Flare V6增强的功能和新特性,包括增强的可视化功能、基于反应的组合库枚举、量子力学 (QM)计算、结合口袋检测和分析、基于AI的小分子自定义扭转参数生成、分子动力学模拟性能增强、FEP预测性能增强、增强的机器学习QSAR方法、增强的GIST水分子位置与稳定性分析、增强的分子对接水分子处理。Flare V6全面的药物发现平台为计算化学家、药物化学家以及学术研究人员分别提供不同级别的灵活软件授权方式。
Flare V6包含了增强的功能和新特性,包括化合物库枚举、量子力学 (QM) 以及口袋检测和分析。Flare V6全面的药物发现平台为计算化学家、药物化学家以及学术研究人员分别提供不同级别的灵活软件授权方式。
增强的3D图形技术
增强了配体和蛋白质表面渲染,以及可调节的光线位置、3D背景效果和增强的景深(图 1),使您更容易充分了解蛋白质-配体复合物的三维结构特征。
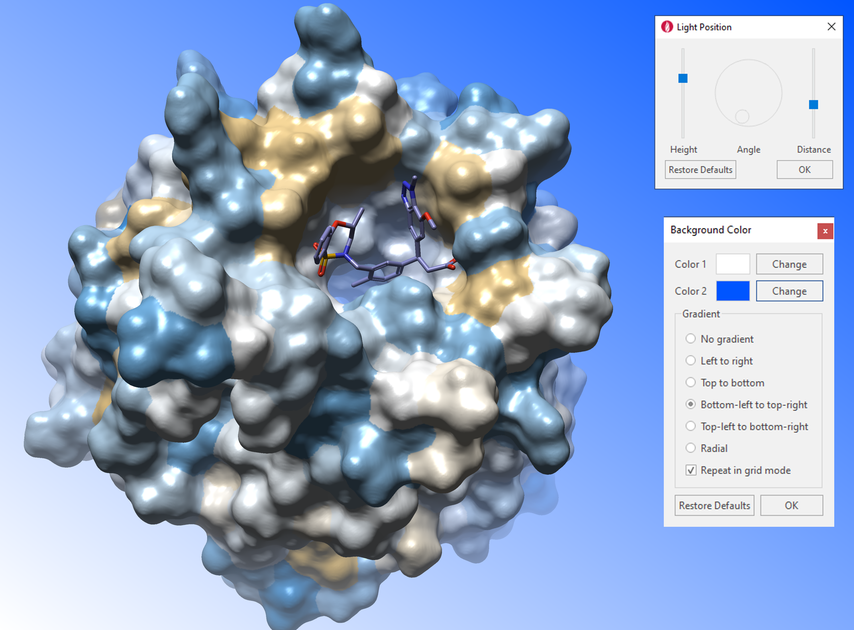
图1. 增强的蛋白质和配体表面、可调节的光线位置和背景效果显着增强了Flare 3D视窗中分子的立体感 (PDB: 5FNU) 。
有利的配体-蛋白相互作用和空间冲突的可视化也得到了改进,相互作用接触可显示为具有可定制颜色的细管,如果需要还可以用开/关选项来控制相互作用接触标签的呈现(图2-左)。 在Flare V6里新增了对疏水相互作用接触的呈现。
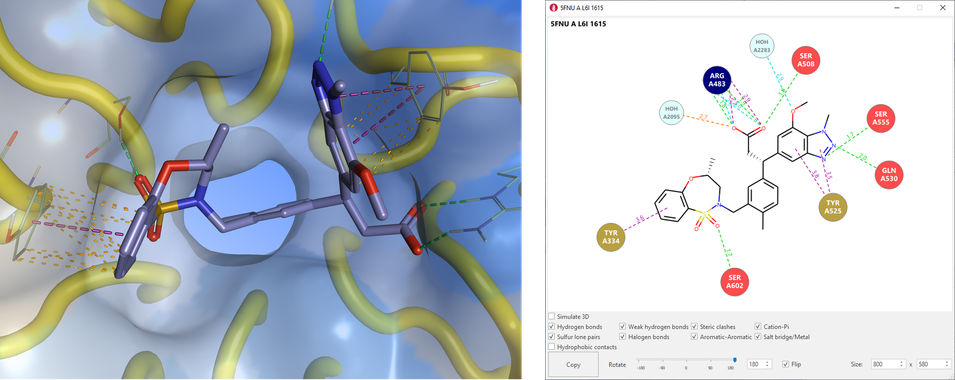
图2. PDB 5FNU的配体-蛋白质相互作用。左:Flare中增强的相互作用显示。颜色可定制:在这张图片中,疏水相互作用默认颜色为橙色;氢键以绿色显示,芳香相互作用以紫色显示。右图:复合物结构PDB 5FNU的2D相互作用图。
新增的2D相互作用图(图 2 右)是显示配体-蛋白相互作用接触的另一种方式,清晰的有利相互作用和空间冲突2D图可以方便地用在演示文稿(PPT)、报告和出版物中。
基于反应的化合物库与阵列枚举
新增了化合物库枚举(Library Enumeration) 模块(图 3)使您能够用50多个常见的合成化学反应来枚举您的化学库或阵列。使用RDKit化合物库枚举的用户会发现:使用Flare用友好界面来进行虚拟反应是多么轻松的一件事。
中小型化合物库可以在图形界面里直接创建,而更大型化合物库的创建可将结果保存到硬盘。在化合物库枚举实验期间可使用性质与子结构过滤器以便将结果聚焦于理想的物理化学空间上。
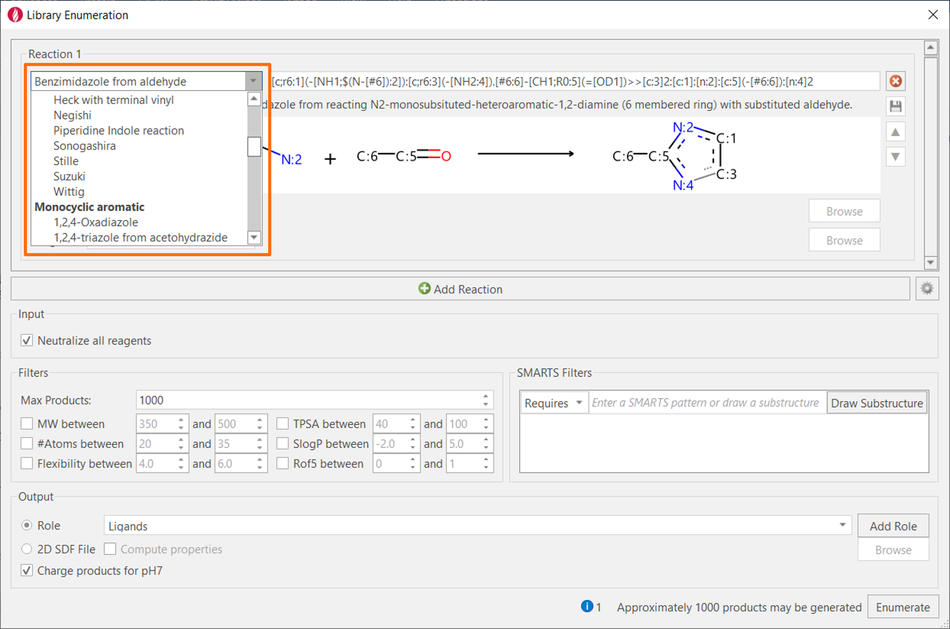
图3. Flare的库枚举包含了常见的50多种化学反应
配体分子的量子力学计算
此版本中还新增了一个基于PSI4实现的配体量子力学 (Quantum Mechanics,QM) 计算模块。您现在可以在Flare中使用QM来对单个配体、构象系综、配体结合构象进行几何优化和单点能量计算,还可以计算和显示配体的分子静电势(图4),对感兴趣配体的特定旋转键进行基于QM的两面角扫描分析(QM Torsion profile)。
如果你担心QM计算速度太慢,还可以通过Cresset Engine Broker™连接到本地集群或云计算设施来加速QM计算、优化计算时间。
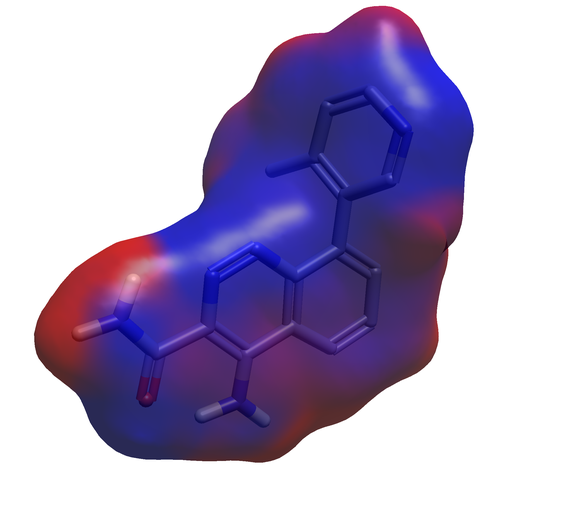
图4. QM提供了对配体电子结构的准确描述,使分子静电势能够在较高的理论水平上进行计算。红色 = 正静电; 蓝色 = 负静电。
结合口袋预测与分析
识别小分子配体潜在的成药性结合位点是新靶点和先导化合物分子建模的关键步骤。Flare中的结合口袋探测和分析可用于搜索单个蛋白质结构中的不同口袋或孔穴,并监测分子动力学模拟轨迹中口袋的打开频率和成药性。结果显示为多色表面(图 5)映射不同类型的潜在结合口袋,每个口袋在口袋信息表中由包括成药性打分值在内的不同参数进行表征。
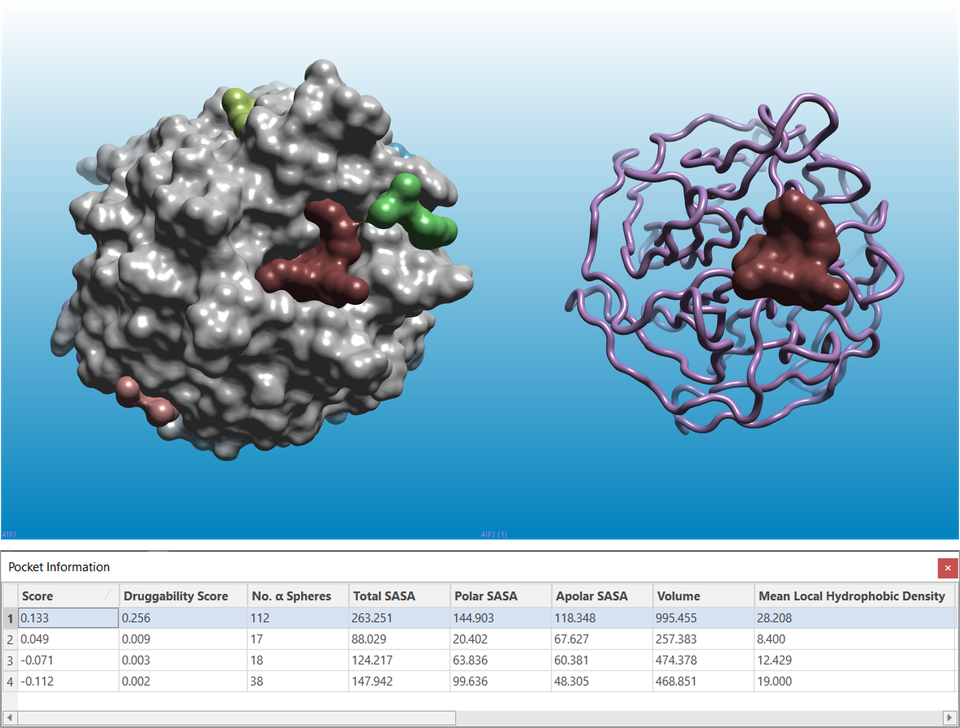
图5. 对KEAP1 (PDB 4IFJ) apo结构进行结合口袋检测的实验结果。左图:KEAP1蛋白的溶剂排除表面显示为灰色。 右图:KEAP1蛋白结构显示为粉红飘带。在两张图片中,口袋都显示为多色表面。口袋信息表(底部)根据不同的参数报告口袋的特征。
基于AI的小分子自定义扭转参数生成
准确的力场参数对于可靠地评估小分子配体和辅酶(cofactor)的热力学性质至关重要。特别是扭转参数(Torsion parameters)至关重要,因为它们描述了分子内最大的局部运动,从而在分子动力学模拟和Flare FEP计算中影响配体的构象分布。
在Flare V6中用基于ANI-2x深度学习QM近似的工作流对小分子自定义扭转参数的计算进行了重新设计(图 6)。这种机器学习势能计算成本只是传统 QM旋转扫描所需的一小部分,却可以达到ωB97X/6-31G(d)密度泛函理论水平的准确性。
使用Cresset Engine Broker™可以在CPU集群上实现完全并行计算,进一步加快自定义扭转参数的生成。
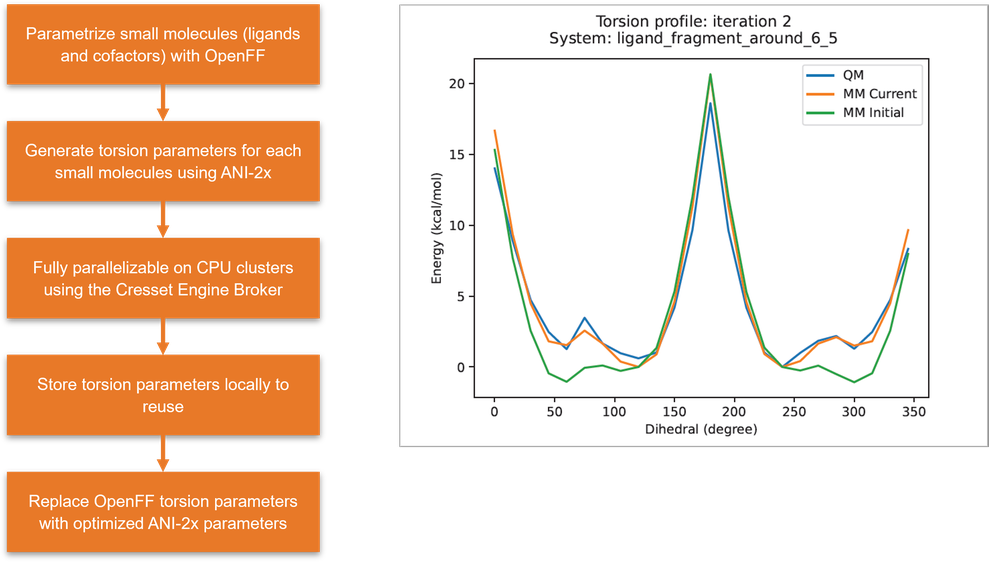
图6. 使用ANI-2x深度学习QM近似为小分子生成自定义两面角力场参数。
分子动力学模拟新特性与分析工具
Flare V6新增了可视化工具以方便分子动力学模拟轨迹的分析和结果的解释。
交互式2D RMSD图(图 7)可用于监测在动力学轨迹中的蛋白质结构变化,使您能够检查分子系统是否处于某种构象状态或重新到达先前探索的状态,并识别帧与帧之间大的构象变化。
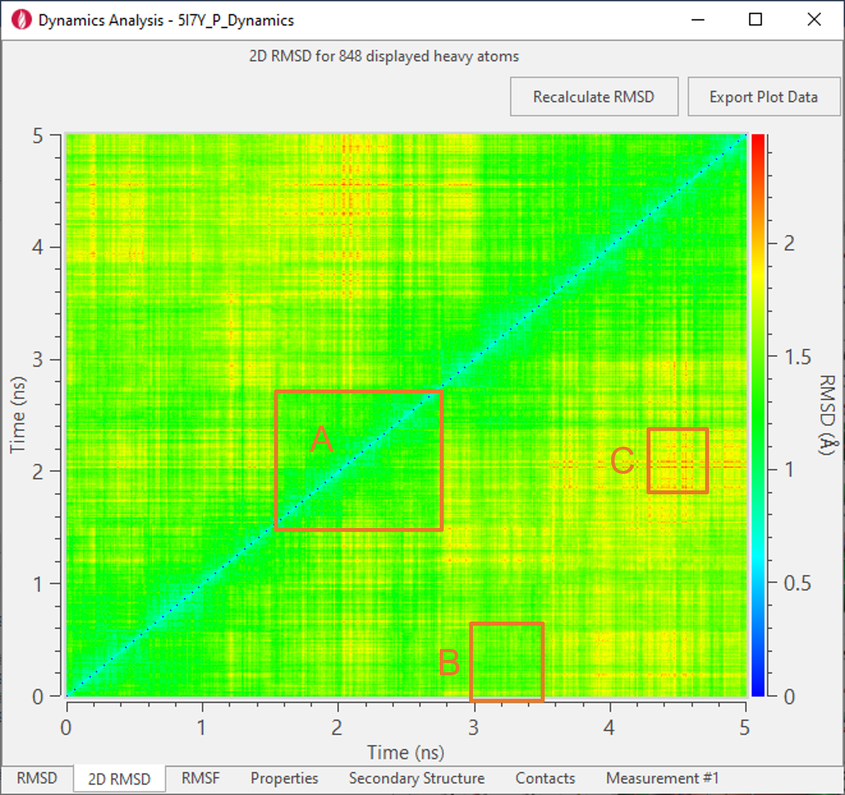
图7. 2D RMSD图可帮助您监测分子动力学模拟轨迹上的蛋白质变化。沿对角线方向低RMSD值(蓝色/绿色)区块(A) 表示给定状态的占用;对角线之外的区块 (B) 表示轨迹正在重新访问较早的状态。 红色单元格 (C) 表示帧之间的大构象变化。
RMSF图可监控单个残基的柔性,指示出在模拟过程中哪些残基对蛋白质运动的贡献最大(图8-左)。二级结构表显示了在分子动力学模拟的每一帧蛋白质中每个残基所采取的二级结构图(图8-右)。

图8. 左图:RMSF图绘制了在整个模拟过程中每个蛋白质残基的均方根波动 (RMSF)。右图:二级结构表显示了在分子动力学模拟过程的每一帧蛋白质中每个残基所采用的二级结构。
还对相互作用接触表进行了增强,该表在动力学轨迹上监测有利的配体-蛋白质相互作用,例如氢键、芳香-芳香相互作用。该表(图 9)现在还可选地监控疏水相互作用,用“条形码”图来呈现每个相互作用接触在轨迹的每一帧中出现或没出现。
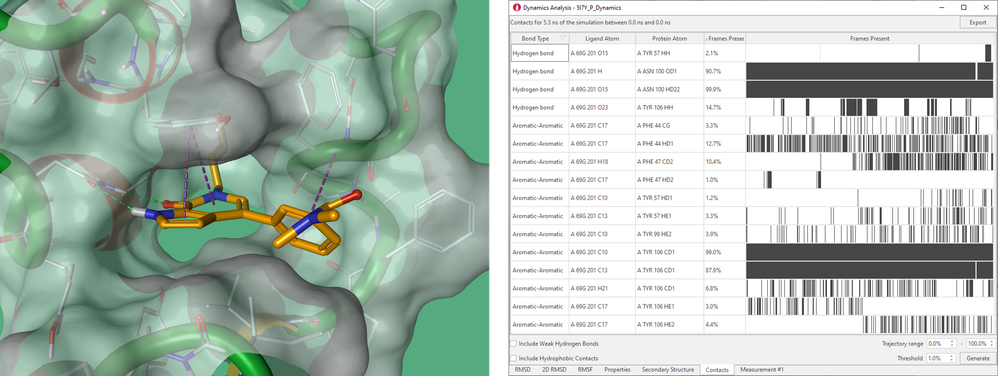
图9. 左图:在PDB 5I7Y配体-蛋白复合物结构短时间分子动力学模拟的第801帧处出现有利的配体-蛋白相互作用。右图:接触表监控着在动力学模拟轨迹中出现的有利的配体-蛋白质相互作用。
此外,改进了系统性质图(势能、温度、盒子体积、密度)以显示性质的移动平均值,以及距离、角度和扭转角的监控绘图现在还可以显示为线型图或直方图。
Flare V6中的分子动力学模拟还包括增强的高级系统选项,显著地扩展了该方法的适用性:
- 磷脂膜的模拟
- 更多的水模型
- 设置模拟的溶剂离子强度
- 在模拟过程中用距离约束来限制大蛋白质的运动
增强的Flare FEP预测性能与新特性
新版本对Flare FEP的核心算法进行了优化,提高了对较大变换的预测性能。这些变化连同使用ANI-2x自动生成自定义扭转参数,提高了Flare FEP的预测性能。
你现在可以更轻松地对FEP实验中有问题的转换进行故障排除。有问题的分子对连接(link)通常与高滞后和/或不良重叠矩阵相关,应进一步研究以评估对模拟条件的微调是否可以改善结果。在理想情况下,因为很难预测新参数是否会比最初的参数得到更好的结果,您希望在不丢失原始结果的情况下重新对分子对转换进行计算。
新增的“复制分子对连接”(Duplicate links)功能(图 10)使您能够针对选定的分子对连接使用自定义的设置将多个实例添加到微扰网络中:例如,手动设置lambda窗口数量和间距,或使用更长的模拟时间。分子对连接的新实例重新计算后,您可以决定是否保留新实例并将结果保存在相对活性计算中,或者是否从结果中将一个或多个实例完全删除。

图10. 使用自定义设置对分子对连接进行多次计算并保持最佳结果,实现对有问题的Flare FEP转换进行故障排除。
在新版本中对Flare FEP的增强还包括新的高级系统选项,例如:
- 更多的水模型
- 新增选项用来设置模拟的温度、压力和溶剂离子强度
- 在模拟过程中用距离约束来限制大蛋白质的运动
先进的QSAR方法
Flare V6新增了强大且经过充分验证的机器学习方法,可用于预测配体的活性和ADMET性质。
其中包括用于构建回归模型的高斯过程(Gaussian Process)方法和用于构建分类模型的随机森林(Random Forest )方法。支持向量机、k-最近邻和随机森林回归仍然可用,“自动”模型构建选项可自动运行所有方法并给出具有最佳统计性能的模型(图 11)。
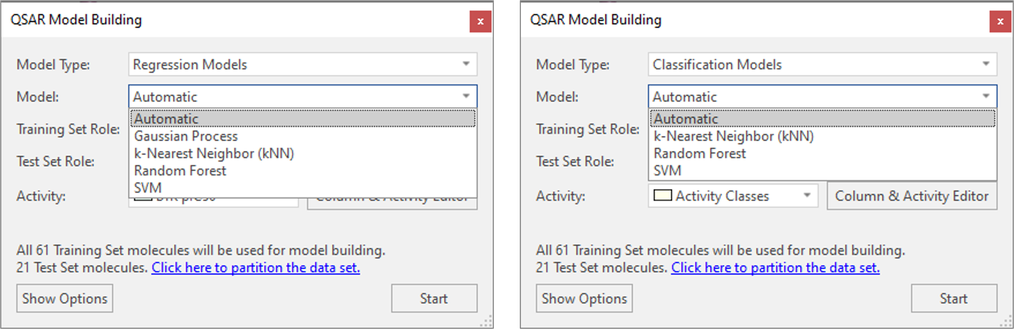
图11. 在Flare V6中强大且经过充分验证的机器学习方法
与以前版本的Flare一样,使用Cresset 3D描述符,叠合配体的静电和形状性质建模,以及自定义2D和3D描述符,使得这些机器学习方法可用于构建稳健的QSAR模型用于预测化合物的活性和ADMET性质。
增强的GIST表面
Flare GIST使用内嵌的OpenMM分子动力学模拟引擎来分析水的位置和稳定性。该方法可以针对空的活性位点(apo 结构)以及配体结合的活性位点(holo结构)蛋白质进行计算:无论哪种情况,它都会生成热表面图,突出显示水稳定性高和低的区域。
Flare V6新增的水分析表面显著提高了GIST结果的可解释性。例如,图12显示了在PDB 4ZLZ上运行GIST水分析实验的结果。在配体-蛋白质复合物中,抑制剂通过桥接水分子(球和棒)与布鲁顿酪氨酸激酶 (BTK) 的p-loop结合。桥接水分子产生三个氢键相互作用,一个与配体相互作用,两个与BTK活性位点相互作用。
GIST分析正确识别出了桥接水分子在PDB 4ZLZ复合物结构中为高水密度区域。在该区域内水分子被预测为焓快乐(enthalpically happy),这一点也不奇怪,因为它可以形成一个很好的氢键相互作用网络。然而,GIST预测该水分子在熵方面是不快乐的(entropically unhappy),从熵的角度来看,水更愿意呆在更加无序的溶剂水中。
由于与桥接水重叠的区域总体上具有正ΔG(在指定阈值处)并且位于水高密度区域,因此将其映射为局部不快乐水(local unhappy water)。位于该区域内的水应该可被模拟桥接水形成的氢键相互作用网络的取代基置换(displace):这与实验证据一致(参见算例PDB 4Z3V)。
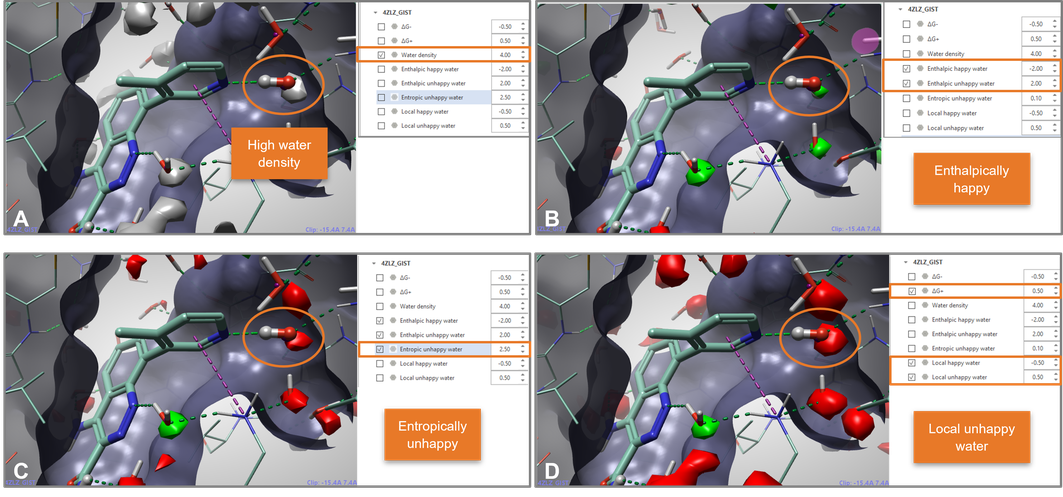
图12. A:GIST 分析正确地将PDB 4ZLZ复合物结构中桥接水分子的位置是被为高水密度区域。B:GIST分析将桥接水分子绘制为焓happy water。 C:预测的熵unhappy water。D:总体而言,GIST预测为局部unhappy 的水应该可以用模拟其氢键相互作用的取代基置换,这与实验证据是一致的。
分子对接与打分时的水分子开关
活性位点中的水分子可强烈地影响到配体-蛋白质的结合。当水分子可置换时,通常会有配体通过桥接水分子与蛋白质活性位点结合,而其它配体则能够直接与蛋白质相互作用。
在Flare V6中,可以将选定的水分子标记为”闪烁(flickering)”,这意味着在对接实验期间可动态地开/关该水分子,分子对接自行决定是否将标记为“闪烁”的水作为蛋白的一部分与之相互作用还是被配体替换,以实现配体的最佳对接模式(图 13)。
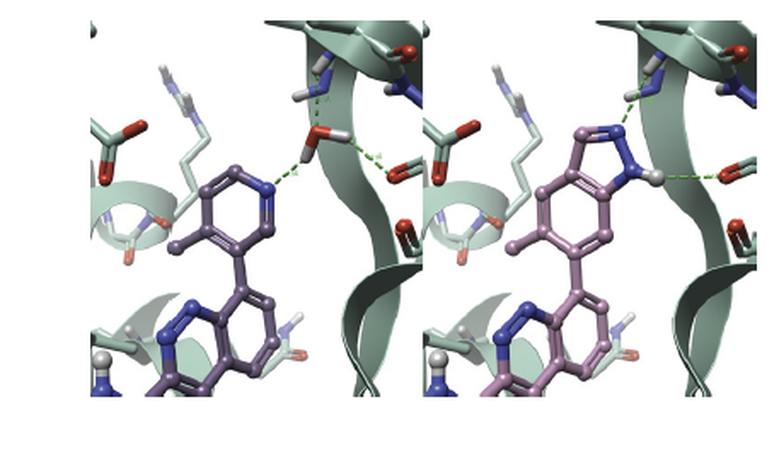
图13. 在对接过程中,可以设定水是否自动打开/关闭,以便为每个配体生成最佳结合模式。
在Flare V6中对接和打分功能的增强还包括:扩展了共价对接的弹头和亲核残基的列表,新增冻结选定的扭转角到所需角度以便在对接期间更好地模拟晶体构象的能力,以及改进的对接和打分图形界面。
灵活的软件授权
无论是计算化学、药物化学用户,无论是工业还是学术用户,总有一款适合你的软件使用授权方式。


