
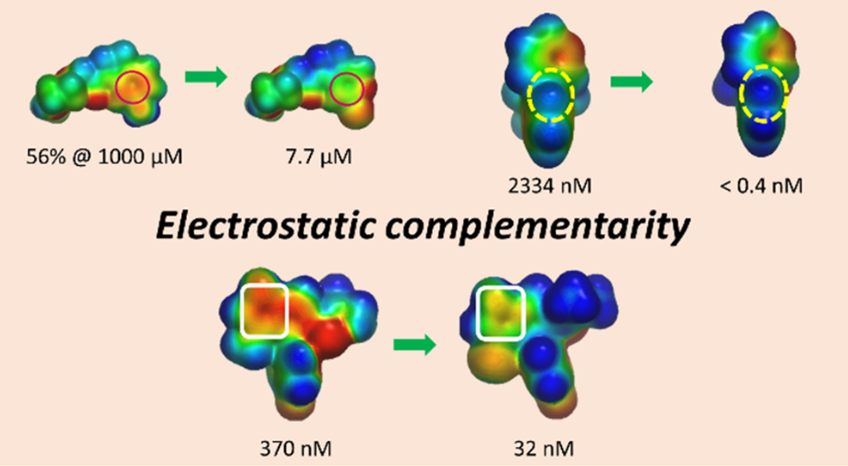
摘要:在基于结构的药物发现中,静电互补性的优化是提高分子对特定蛋白靶标亲和力的重要策略。在本文中,作者列举了几个优化蛋白-配体静电互补性或分子内静电相互作用以改善靶标亲和力、物理化学特性、体外性质和脱靶选择性的例子。作者还回溯性地用静电势表面互补性分析方法研究了一系列活性差异高达8000倍的FXa抑制剂。使用图卷积深度神经网络可快速生成高质量的ESP表面,这使得在药物化学领域中广泛地使用静电互补性分析这种工具成为可能。
原文:Cons, B. D.; Twigg, D. G.; Kumar, R.; Chessari, G. Electrostatic Complementarity in Structure-Based Drug Design. J. Med. Chem. 2022. https://doi.org/10.1021/acs.jmedchem.2c00164.
编译:肖高铿/2022-06-06
1. 简介
静电相互作用在分子识别中起着关键作用,其特征和调节是基于结构药物设计(Structure-Based Drug Design, SBDD)的核心。在SBDD中,最大限度地提高配体和蛋白质之间的形状和静电互补性是提高结合亲和力和反靶选择性的公认成熟策略。然而,静电互补性(electrostatic complementarity, EC)是一个比形状互补性更复杂的概念。
静电相互作用的能量和优势方向并不容易评估,即使是富有经验的药物和计算化学家也是如此,而对去溶剂化的惩罚加剧了这项任务的复杂度。
静电势(Electrostatic potential,ESP)表面的计算有助于这项任务,一方面可以识别静电匹配或不匹配的区域,另一方面可以方便地比较具有不同取代物或母核骨架的多个配体。例如,人们已经对在芳香体系中取代基对ESP的影响进行了广泛研究,不同程度地归因于π-共振、σ-诱导和静电场效应1-4,而芳香π-体系的总电子密度的变化对之影响较小4,5。ESP表面的使用考虑了这些因素,可用于快速理解蛋白质-配体的互补性。
在本文中,我们总结了一些SBDD案例,其中ESP分析和随后的调整用来优化蛋白-配体的静电互补性,以提高活性和选择性。随后,我们还讨论了如何应用ESP方法来调节分子内的相互作用以稳定配体的生物活性构象。
2. 静电势的计算
分子静电最广泛的研究方法是对分子ESP表面的可视化与分析6。分子表面任意给定点的ESP能量是分子电荷分布和位于该表面点的单位正电荷之间的相互作用能。ESP可以通过量子力学(quantum mechanical, QM)从头计算(ab initio)的方法进行精确地计算,由于对复杂体系需要昂贵的机时,该技术主要限于对小分子的计算。例如,图1B显示了用ab initio密度函数理论(Density functional theory, DFT)计算的氯苯ESP表面。此外,连续溶剂模型可用于QM方法以模拟溶剂对静电相互作用的影响7-9。为了提高计算速度,对原子中心电荷进行了参数化以重现QM方法得到的ESP表面10-16。这些电荷可用于分子力学力场并用于计算ESP表面6;例如,图1C显示了使用AM1-BCC原子中心电荷生成的氯苯ESP表面17。因为使用了预定义的原子类型,相同的原子电荷可以用于不同的物种,所以这种原子电荷可快速地计算蛋白质和DNA等大分子的ESP。为了给原子中心电荷建立连续溶剂模型,人们开发了快速解释泊松-波尔兹曼方程(Poisson− Boltzmann equation)的方法,并在诸如微管和核糖体之类的复杂大分子上证明了可用性18-23。
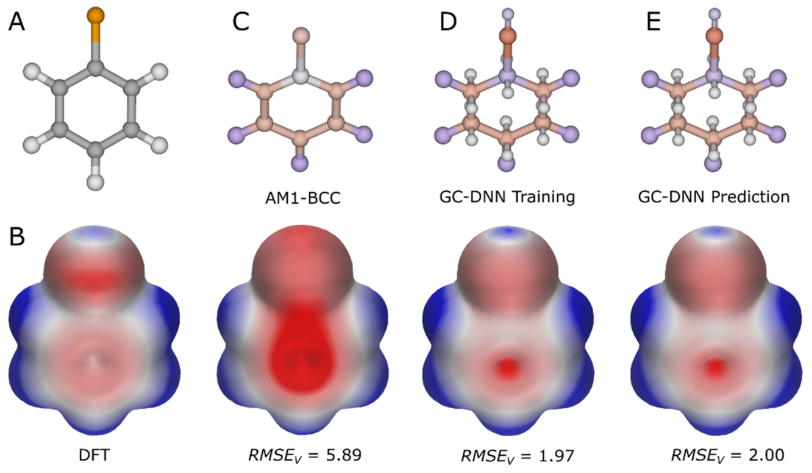
图1. 用各种方法得到的氯苯ESP。(A)氯苯的化学结构,灰色球体:碳原子,橙色:氯原子,白色:氢原子。(B)用ab initio DFT B3LYP方法计算的ESP表面,作为ESP表面比较的标准,红-白-蓝颜色梯度代表值为-15~15kcal/mol的ESP。(C)根据AM1-BCC偏电荷值对原子进行着色,红-白-蓝颜色梯度代表-0.5到+0.5的电荷(顶部),以及根据这些电荷计算的ESP表面(底部),RMSEv体现了ESP能量相对于从头DFT ESP能量的偏差。(D)代表p轨道和σ孔的原子偏心电荷用连接到碳原子和氯原子的小球表示。原子/特征根据偏电荷值着色,优化这些电荷值以重现从头算的DFT ESP表面。使用这种拟合电荷计算的ESP表面显示在底部。较低的RMSEv值表明原子偏心电荷提高了ESP表面的准确性。在本研究中,这些电荷用于训练图卷积深度神经网络(GC DNN)模型[24]。(E)用GC DNN模型预测的电荷(顶部)以及使用该电荷计算的ESP表面。该种方法计算ESP表面与DFT拟合的原子/特征电荷的精度几乎相同,而无需要耗时的从头DFT计算。
虽然使用原子电荷可以更快速地计算ESP表面,但与QM方法得出的ESP表面相比,其精度较低,如图1C中的均方根误差值(root-mean-square error value,RMSEv)所示。使用原子电荷的主要限制是没有σ孔(σ holes)、π轨道(π orbitals)和孤对电子(lone pairs)等特征,这些特征要么合并在原子中心,要么在参数化过程中没被考虑到25-28。图1D将氯苯的π轨道和σ孔呈现为原子偏心的特征点(用连接到碳原子和氯原子的小球体表示)。一些力场,比如XED,明确地将这些原子偏心的位点作为额外的电荷中心以提高精度,更好地描述了电子各向异性系统29。用XED力场计算的ESP已用于开发SBDD工具来评估蛋白-配体复合物的静电互补性30。
另一个主要限制是由于在电荷参数化过程中使用的分子数量有限(特别是于小分子)导致原子电荷地不正确分配。出于实用的目的,QM方法多用于计算小分子的ESP,而原子电荷多用于计算蛋白质的ESP。然而,由于高昂的计算时间成本(分子量为400的典型类药分子的QM ESP计算大约需要30分钟),QM方法对于交互式药物设计应用仍然是一个瓶颈。
为了克服用经验模型计算电荷的问题,人们开发了各种机器学习/深度学习方法来建立预测电荷的模型31-37;这些方法可以用于分子力学计算和分子动力学模拟。当σ孔、π轨道和孤对电子等特征作为原子外位点考虑进去的时候,其推导出的电荷(通过最小二乘法拟合)可重现QM ESP,计算的ESP表面与从头算的ESP表面非常相似(图1BD)。
最近,Rathi等人通过拟合100000多个分子的QM ESP获得原子和原子偏心位点的电荷,训练和验证了一个图形卷积深度神经网络(GC-DNN)模型24。一组测试化合物的预测电荷被进一步用来计算它们的ESP表面,其与相应的QM ESP平均偏差为3.27±0.62kcal/mol(RMSEv),比起AM1-BCC模型得到的平均偏差(RMSEv=7.86±1.04kcal/mol)有了显著的改善。图1C、E中的氯苯可作为示例说明。此外,作者还实现了一个经验模型,将带电分子中的带电原子的偏电荷(partial charge)按比例缩小,以进一步改善预测ESP表面的可视化。虽然这种按比例缩放限制了不带电和带电分子ESP之间的直接比较,但它仍然允许在一系列带电分子中进行相对比较,以达到在SBDD中优化蛋白质-配体相互作用的目的。总的来说,该模型能够在适合交互式SBDD的时间尺度上计算出高度精确的配体和蛋白质ESP表面(对于分子量为400的典型类药分子的GC-DNN ESP计算大约需要0.3秒),Astex公司的药物化学家们现在已经开始使用这种EPS计算方法。
3. 优化蛋白-配体的静电互补性
3.1 Cathepsin S/L(CatS/L)双重抑制
作为罗氏对小分子药物发现的计算方法综述的一部分,Stahl等人38报道了使用蛋白ESP开发CatS/L双重抑制剂的算例。 罗氏先前报道的一系列选择性CatS化合物作为CatS/L双重抑制剂的起点39,对这两种蛋白ESP表面的分析表明:CatL结合位点具有优势的负表面静电势,在结合位点半径7Å内有6个酸性残基Aps与Glu而没有碱性残基Lys和Arg;而CatS结合位点在相同的半径内却仅包含一个酸性残基Asp和一个碱性残基Lys(图2)。

图2. (A) 映射了静电势的人类CatL结合位点 (左, PDB 2yjc) 和 (B) 映射了静电势的小鼠CatS 结合位点(中, PDB 4bpv) 。在化合物1附近7Å内发现以下带电氨基酸:人类CatL(2 Glu,4 Asp),人类CatS(1 Asp,1 Lys),小鼠CatS(1 Glu,1 Asp,1 Lys)。 (C) 化合物2与人类CatL (PDB 5f02) 的X-衍射共晶结构。使用内部开发的代码库 (https://github.com/AstexUK/ESP_DNN/tree/master/esp_dnn/ext/pli) 来添加、优化蛋白质和配体中的氢原子。使用GC DNN模型生成ESP,以-50(红色)到 0(白色)到50(蓝色)kcal/mol的颜色梯度映射到实体的Connolly分子表面24。
为了改善与CatL的静电互补性,将化合物1中的环丙基连接臂换为一个碱性的氮杂环丁基(图2)。这使得对CatL的结合亲和力提高了11倍,而对CatS的亲和力只降低了2倍。化合物2还保留了对Cathepsin K(CatK)的良好选择性窗口。化合物2与CatL的X-衍射共晶结构如图2c所示,氮杂环丁基与酸性残基没有直接的相互作用,但由于静电相互作用的长程性质,活性提高的益处是肉眼可见的。基于先前报道的化合物40对CatL S3口袋卤键相互作用的探索,对5-氯吡啶的进一步优化得到了化合物3,一种对CatL和CatS具有单位数nM级别的抑制剂,并保持了对CatK的选择性(图3)40。
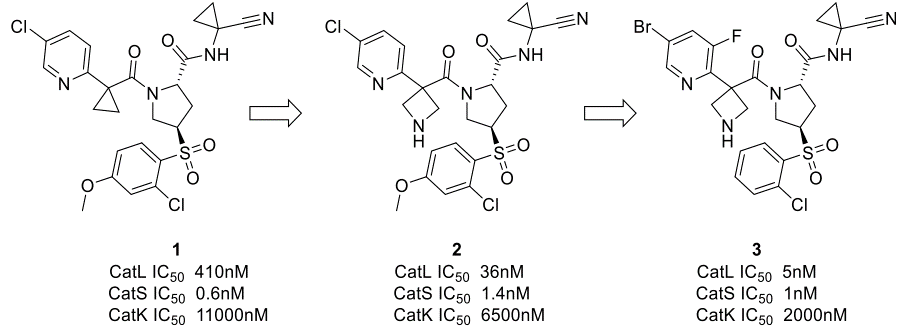
图3. 将CatL抑制剂1优化为CatL/CatS双重抑制剂3。
3.2 Factor Xa(FXa)抑制剂
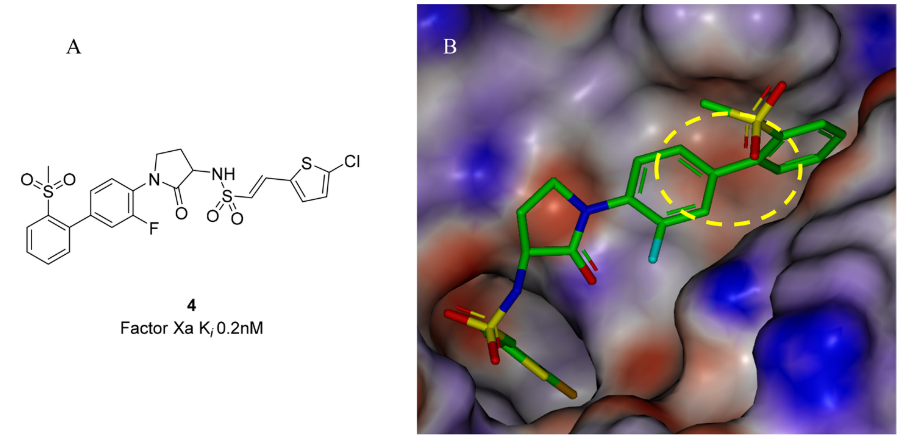
图4.(A)FXa抑制剂4的结构。(B)4与FXa的复合物(PDB 2VH6)。蛋白和配体中的氢按照图2所述的方法进行了添加和优化。蛋白的ESP表面用GC-DNN方法计算并以-50(红色)到0(白色)到50(蓝色)kcal/mol的颜色梯度显示在实心Connolly分子表面上。
2008年,葛兰素史克公司披露了一系列FXa抑制剂的优化结果41。先导化合物4与FXa的共晶结构表明,芳基砜结合在S4口袋中,与Trp215形成边对边的芳香相互作用。芳基砜的邻位氢靠近Trp215的富电子吲哚环(图4中的黄色椭圆形高亮处),因此作者假设,这种相互作用及抑制剂的亲和力可以通过另一个邻位(4中的甲基砜)取代基的σ电子吸电效应来调节42。
表1. 化合物5-15与FXa结合亲和力

在寻求优化S4口袋相互作用的同时,也希望降低化合物的亲脂性,因此用吡啶取代了化合物4中的邻-氟苯基,并在末端芳香环的邻位上引入了一系列取代基得到了化合物5-15(见表1)。将这些化合物的亲和力对Hammett σm系数43作图(图5A)可确认Hammett σm系数与FXa亲和力之间具有明确的关系。计算的趋势线表明,根据Hammett σm系数可以预测未知化合物的亲和力(pKi = 5.28σm + 6.39,R2=0.897)。
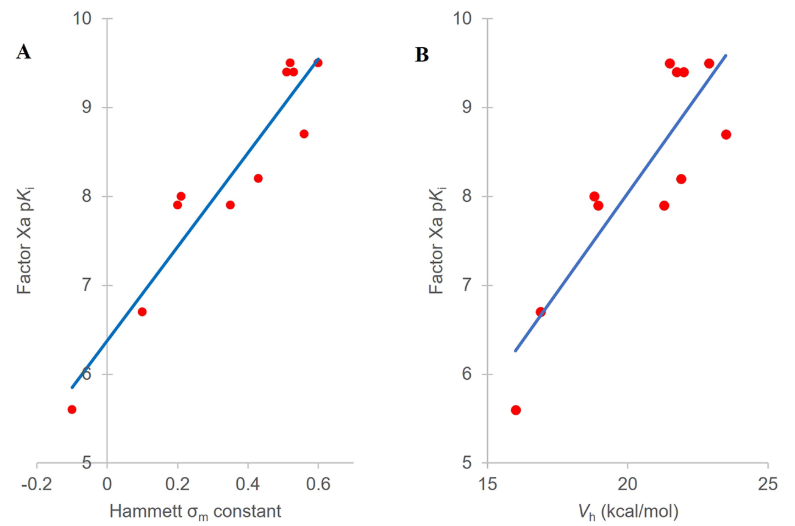
图5. (A) 化合物5-15的pKi与Hammett σm常数之间相关性。化合物5-7的pKi值是基于可能的最大亲和力计算,化合物5为0.4nM,化合物6、7为0.3nM。(B) FXa因子pKi与化合物5-15末端芳香环上邻位氢上计算电荷(Vh)之间的相关性,其中电荷是采用GC-DNN模型来计算的。
虽然本文描述的优化工作需要合成所有列出的化合物,但用ESP计算有可能预测出活性最强的分子从而减少需要合成的化合物数量。为了说明这一点,用GC-DNN ESP模型计算了S4口袋结合片段联芳基的ESP表面(见图6的化合物5、12和15算例)24,结果发现,邻位氢的电荷(Vh)可以正确地对化合物的亲和力进行排序(见图5B,R2 = 0.757)。这个例子证明了ESP计算的性能:在初步确定了SAR的情况下,可以准确地预测化合物的活性,类似的方法可以应用于各种靶标蛋白。
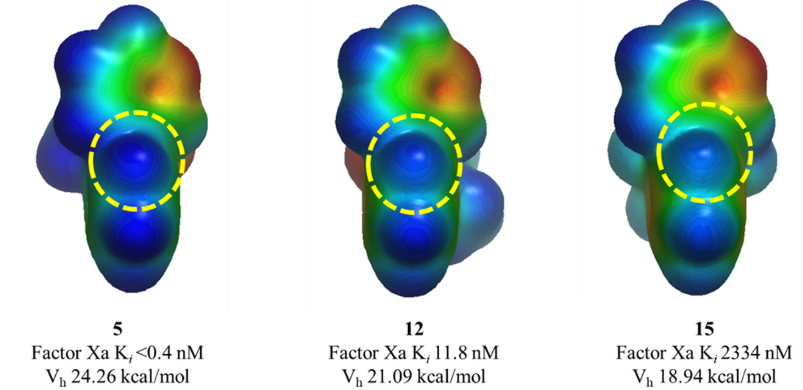
图6. 化合物5、12以及15的联芳基S4口袋取代基的ESP表面。与Trp215相互作用的邻位氢以黄色虚线圆圈突出显示。Vh是虚线圆圈中邻位氢上的点电荷。ESP和点电荷用GC-DNN计算并以-50(红色)到 0(绿色)到 50(蓝色)kcal/mol的颜色梯度显示在表面上[24]。
3.3 凋亡蛋白抑制剂(Inhibitors of Apoptosis Proteins,IAP)
2015年,Astex的科学家们报告了针对细胞凋亡抑制蛋白1和2(cIAP1/2)和X-linked抑制凋亡蛋白(XIAP)的片段发现和优化[44]。在针对XIAP的杆状病毒IAP重复3(BIR3)结构域的片段苗头化合物中,优先跟进片段16。通过使用SBDD和一个小型的集中库,将吡咯烷17优化为二氢吲哚18(图7)。
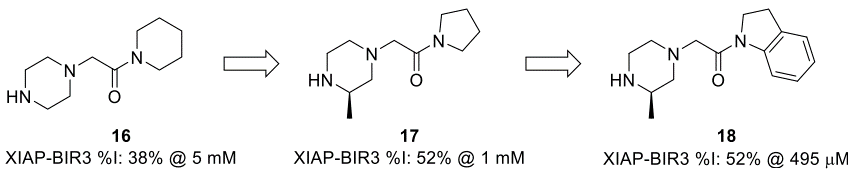
图7. 16-18片段的优化
虽然该变化仅有轻微地改善活性,但X-衍射晶体结构显示其与蛋白表面有良好的形状互补性,并且有望延伸进入相邻的子口袋以确保获得进一步的优化。为了提高化合物18的结合亲和力,对结合位点的静电环境进行了检查(图8)。ESP表面显示出在化合物相邻的蛋白表面有一个负的静电势互补区,这是由一个骨架羰基和Tyr324的酚羟基氧造成的。X-衍射晶体结构表明,二氢吲哚的芳香部分堆积在这个负静电势之上。
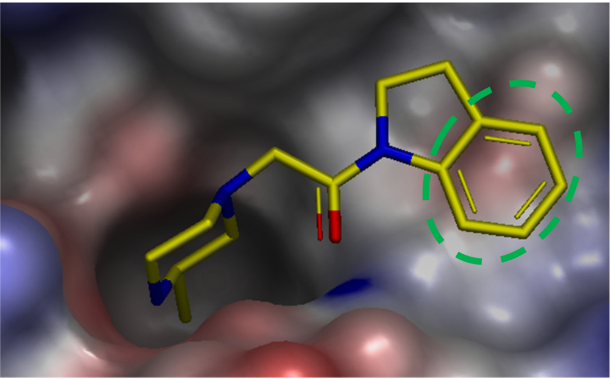
图8. XIAP的ESP表面叠合到化合物18与XIAP-BIR3的X-衍射晶体结构上。绿色虚线椭圆表示负静电表面互补区。蛋白的Connolly表面用ESP以-50(红色)到 0(白色)到 50(蓝色)kcal/mol的颜色梯度进行着色。蛋白和配体中的氢原子按图2所述方法进行添加与优化。
作者假设化合物18对靶标亲和力弱是由于二氢吲哚的负静电π-云和蛋白的负静电表面的静电排斥引起的。为了验证这一假设,合成了一系列电负性不同的化合物18的类似物,根据取代基的Hammett σp值,在二氢吲哚的C-6位置接上精选的给电子取代基或吸电子取代基(表2)45。如图9A所示,化合物的XIAP-BIR3结合亲和力对二氢吲哚π-系统的电子特性非常敏感:比之富电子的二氢吲哚(19和20),缺电子的二氢吲哚(26和27)的活性大约高出两个数量级。
表2. 二氢吲哚类化合物18-27对XIAP-BIR3域和cIAP-BIR3域的亲和力
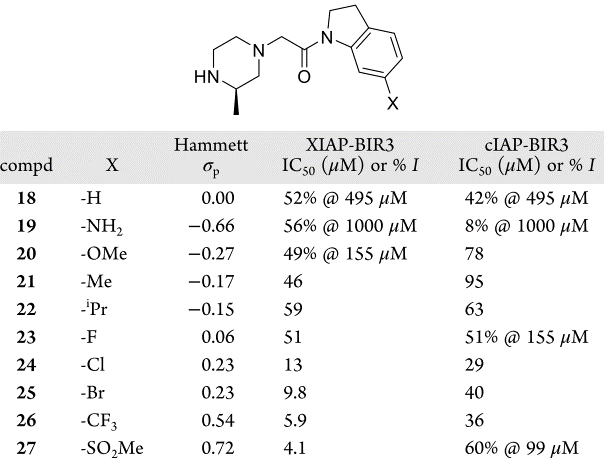
在相关的蛋白cIAP1中,将324位的酪氨酸替换为苯丙氨酸。这导致在XIAP中看到互补的负静电势区减少。当测量18-27对cIAP1的亲和力时(表2),亲和力显示出与Hammett σp值没有相关性(图9B),这进一步证实了在二氢吲哚环和蛋白表面之间的静电互补性优化是化合物对XIAP亲和力增加的原因。
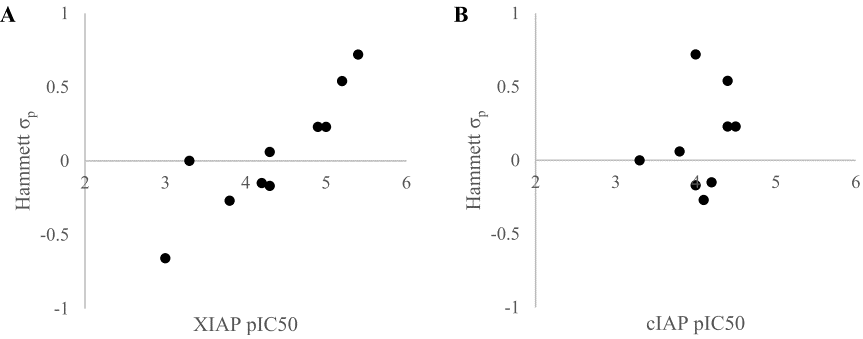
图9. 二氢吲哚类化合物18-27与(A) XIAP-BIR3结构域、(B)cIAP-BIR3结构域的Hammet σp系数对亲和力的相关性(化合物19未在图B中显示)
为了进一步降低二氢吲哚环的正电性,用ab initio方法计算了氮杂二氢吲哚类化合物28的ESP表面,并与二氢吲哚类化合物19的ESP表面进行比较(图10)。结果表明,芳香环的电正性显著降低。合成化合物28,与化合物19相比,它对XIAP-BIR3的亲和力(IC50=7.7μM)提高了约50倍,同时也为进一步细化这些化合物提供了有用的合成处理方法。
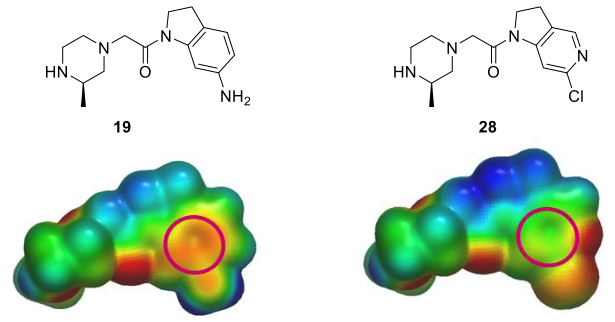
图10. 化合物19、28的ESP表面比较,其中ESP表面以-50(红色)到0(绿色)到50(蓝色)kcal/mol颜色梯度进行着色。红色圆圈表示与XIAP P3口袋中负静电势互补区接触的芳香表面。
3.4 着丝点相关蛋白E(Centromere-Associated Protein E,CENP-E)
在上述讨论的例子中,蛋白-配体复合物的X-衍射晶体结构用于ESP表面的可视化;然而武田制药(Takeda)的Hirayama及其同事发表了一个关于优化着丝点相关蛋白E(CENP-E)抑制剂的例子,其中用基于驱动蛋白纺锤体(kinesin spindle protein,KSP)的运动结构域的同源模型来生成ESP数据46-47。从HTS命中的噻吩并吡酮类化合物29开始,对酰胺的优化发现了化合物30,其生化亲和力有了显著的改善(图11)。在同源模型中对化合物29的分析表明,吡啶酮羰基没有与蛋白发生任何相互作用;这使得作者重新设计母核骨架,得到了噻吩并吡咯类化合物31,它在生化试验中的IC50 = 32 nM。
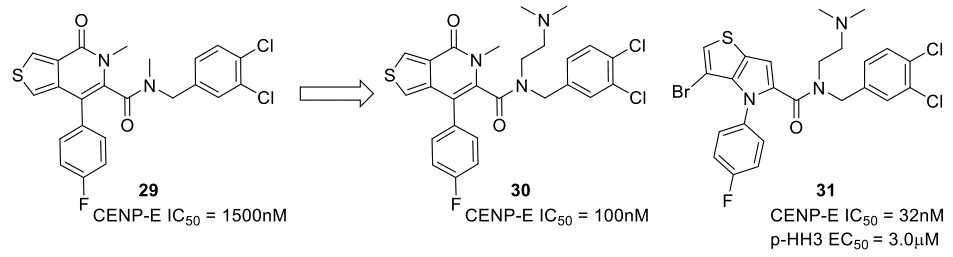
图11. 苗头化合物29的优化
为了更好地理解在噻吩并[3,2-b]吡咯的C-3位上取代基的SAR,计算了一系列类似物的ESP表面(图13)。为了简化计算,将酰胺替换为简单的N,N-二甲基酰胺。诸如3-Br(化合物32)或3-Cl(化合物33)之类的强抑制剂,它们的噻吩上显示出中性的静电势(图12)。相反,3-甲基衍生物34显示出负的ESP,3-腈基类似物35在噻吩环上显示出正的ESP。同源模型表明,该骨架插入到一个由异亮氨酸、酪氨酸、苯丙氨酸和蛋氨酸等残基组成的疏水腔中;因此,作者推测,与CENP-E结合需要一个中性的骨架,这一点得到生化测试数据的证实。
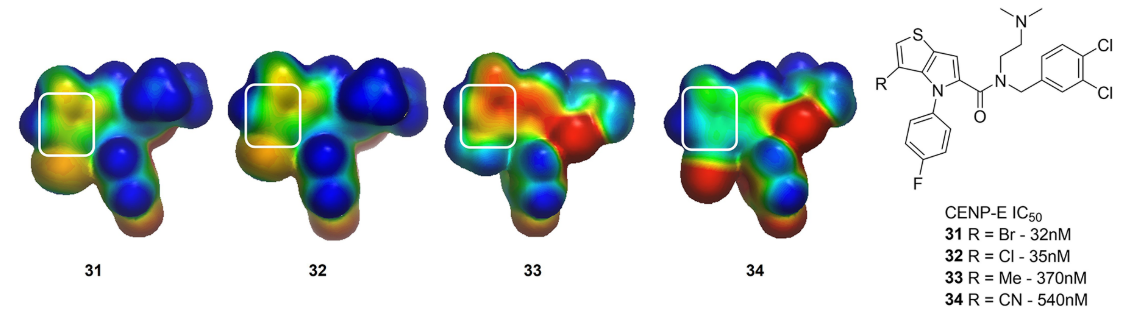
图12. 用ESP理解化合物31周围的SAR。计算二甲基酰胺类似物的静电势以减少计算负担。感兴趣的区域以白色圆角矩形高亮显示。ESP用GC-DNN 模型生成并显示为Connolly表面,颜色梯度为-50(红色)到0(绿色)到50(蓝色)kcal/mol24。
虽然化合物31显示出良好的生化亲和力,但该化合物显示出较差的细胞活性(p-HHE EC50 = 3.0 μM)。为了提高细胞活性,作者尝试对噻吩并吡咯母核进行二次骨架跃迁。根据之前的ESP分析结果,生成了潜在骨架的ESP表面,由于预测到溴代吲哚(化合物36和38)在噻吩环上有一个负的ESP,所以没有合成这些骨架(图13)。预测到咪唑并吡啶类化合物40具有中性的ESP,因此选中该类化合物并进行合成。令人欣慰的是,化合物41显示出与31相似的生化亲和力(50nM对32nM),并且细胞亲和力提高了2倍(1.54μM对3.0μM),同时在PAMPA(pH7.4)试验中测量的渗透性也有显著地改善(化合物31与41的渗透性分别为41nm/s与353nm/s)。虽然细胞亲和力的变化不大,但这些结果为进一步开发这类先导分子提供了信心。
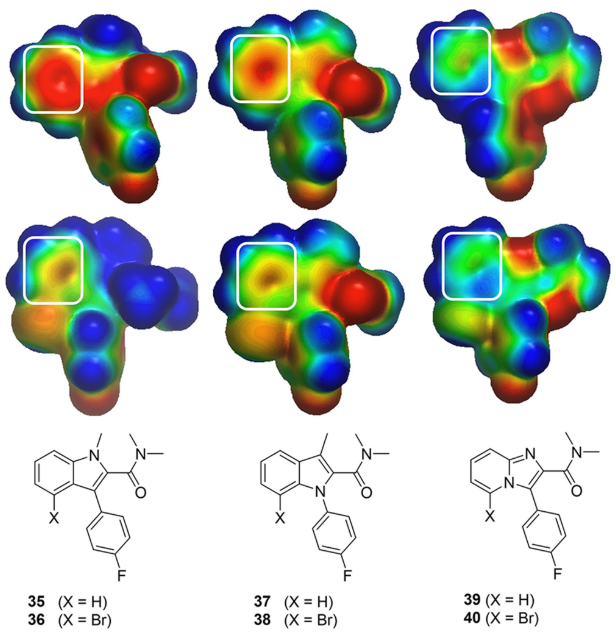
图13. (A) 用ESP表面来预测杂环骨架跃迁以改善细胞的亲和力。正静电势、中性和负静电势区域分别用蓝色、绿色和红色着色以-50(红色)到0(绿色)到50(蓝色)kcal/mol的颜色梯度映射到实心Connolly 分子表面上,其中ESP用GC DNN模型生成24。感兴趣的区域用白色圆角矩形高亮显示。(B)骨架跃迁产生的化合物41。
4. 优化分子内静电相互作用
ESP不仅可以在SBDD中用来优化蛋白-配体的相互作用,还可以用来研究分子内相互作用。单独分析小分子配体的静电特征可以产生有益和有害的分子内相互作用信息,从而使现有SAR合理化,并为前瞻性地设计结合亲和力增强的目标分子提供了机会。。
在SBDD的背景下,与配体非结合构象相比,分析配体结合构象(即生物活性构象)的分子内相互作用具有特殊意义。在药物发现活动中研究的大多数小分子在溶液中游离时具有多种构象,主要由可旋转键的数量决定。溶液中的优势构象和观察到的结合构象之间的密切对应有助于最大限度地减少配体结合时的熵惩罚(熵损失),因此稳定化溶液中的生物活性构象提供了一种增加结合亲和力的安全策略48-49。一维和二维核磁共振光谱是典型的最低能游离配体构象实验测定方法,其中质子化学位移信号和空间相互作用可用于评估配体的柔性程度并归属主要的构象50-51。
柔性配体结构在溶液中的预组织可以通过一系列方法实现,如改变官能团、大环化或引入分子内相互作用38,52-54。根据我们的经验,在后一种情况下,已经证明ESP特别有用,仔细分析在X-衍射共晶结构中观察到的分子内相互作用可以获得理性的设计思路:尝试在主要的溶液构象中再现这种分子内相互作用。下述例子以ESP驱动的π-π堆积和偶极-偶极相互作用的优化为特色,但这种方法也可以应用于其他诸如阳离子-π、疏水或氢键相互作用之类的分子内接触。
4.1 Murine Double Minute 2 (MDM2)
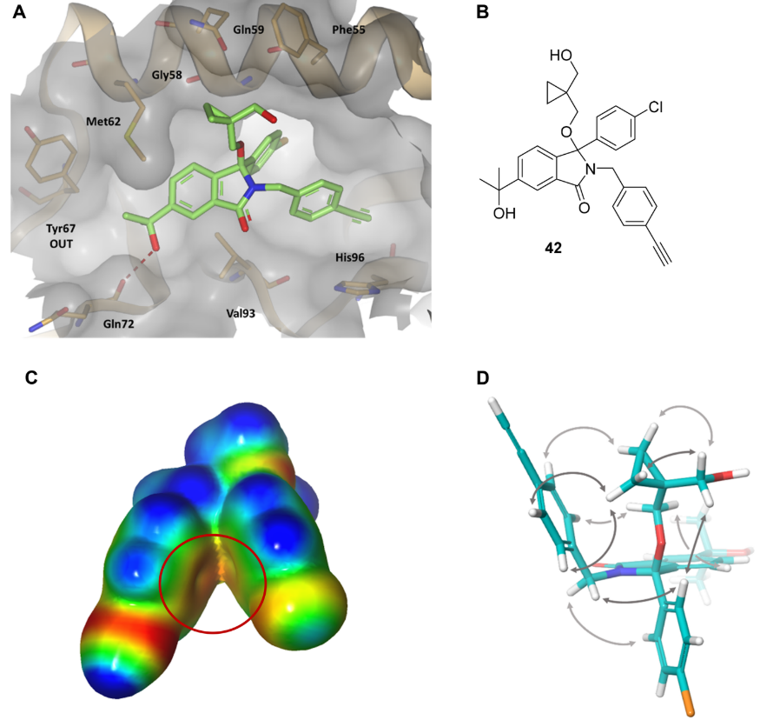
图14. (A) 化合物42与MDM2 (PDB 7BMG) 结合的X-衍射晶体结构。(B)化合物42的结构。(C)化合物42结合构象的ESP表面。负值区为红色,正值区为蓝色。红色圆圈表示芳环的π-系统之间可能存在静电冲突。 (D) 由NMR法确定的42溶液构象。来自NMR谱的空间ROESY相关性用箭头表示。图片来源于J. Med. Chem. 2020, 64 (7):4071-4088,经 Chessari等人许可转载,版权归美国化学学会所有。
在新型异吲哚啉酮MDM2抑制剂的结构引导设计中采用了分子内ESP分析55。早期蛋白质-配体X-衍射晶体结构,例如化合物42(图14A)的共晶结构,表明其中4-氯苯基和4-乙炔基苄基相互堆积形成了一个折叠构象。确定了这种分子内相互作用后,计算化合物42结合构象的ESP表面。结果表明,两个芳环之间存在静电排斥,如图14C所示:两个红色的π-系统ESP表面的非常接近。该理论得到溶液核磁共振研究的支持,研究表明化合物42的优势构象是开放形式,两个芳环相互远离以避免不利的静电冲突(图 14D)。
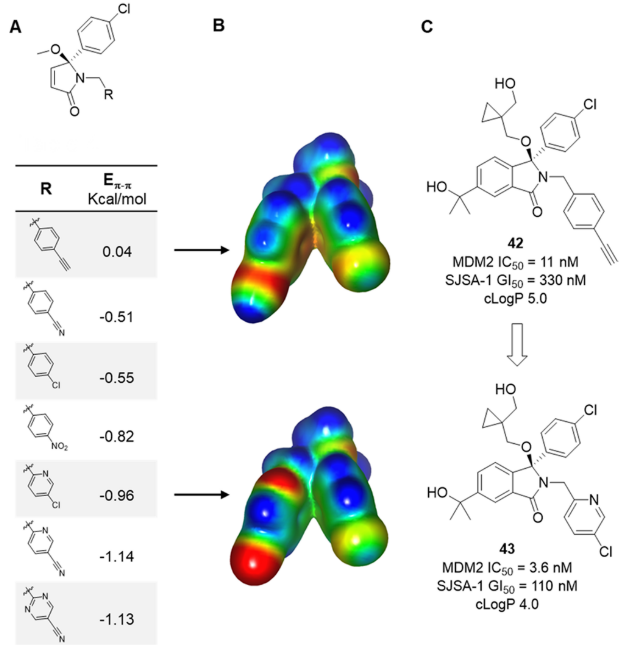
图15. (A) 芳香相互作用 (Eπ-π)的计算值。 (B) A中两个代表性示例的ESP表面。负值区域为红色,正值区域为蓝色。(C)改善化合物42、43的结合亲和力与细胞活性,降低计算的亲脂性。图片来源于J. Med. Chem. 2020, 64 (7):4071-4088,经 Chessari等人许可转载,版权归美国化学学会所有。
在确定了生物活性和溶液构象之间的差异后,进一步用ESP分析以及”热力学循环”方法56来模拟4-乙炔基苄基的潜在替代物,其有利于形成在结合结构中看到的折叠构象(图15)。对两个环之间相互作用能(Eπ-π)的定量分析和对ESP表面的定性检查都表明:越是缺电子的环系越具有明显优势,如图15所示的氯吡啶例子(化合物43)。令人欣慰的是,这一理论得到了证实,因为几种合成的带有缺电子的苄基型取代基类似物在初步的结合试验和SJSA-1 TP53WT细胞试验中均显示出活性的改善,同时亲脂性降低,这有助于控制物理化学性质。例如,与化合物42相比,化合物43在两种试验中的活性都提高了3倍,cLogP下降了1个对数单位。虽然这种活性的改善比上述的例子要小,但这也可根据计算的能量差异所能预期到的。这种方法还允许将极性杂环引入以嵌入到口袋中,该口袋先前需要更亲脂基团的从而将亲脂配体效率(LLE 或LipE)从2.9增加到4.457。化合物43与蛋白的X-衍射共晶结构和单独小分子X-衍射单晶结构证实,正如ESP建模所预测的那样,极性更强的芳香基团可保留其生物活性所需的折叠构象。这个例子说明了ESP具有识别可能的分子内冲突的能力,然后可以帮助理性设计新的目标分子,这些分子在溶液中具有有利的相互作用构象,从而交付活性得以提高的化合物。
4.2 凋亡蛋白的抑制剂(IAP)
表3. 化合物44、46-47的结合亲和力
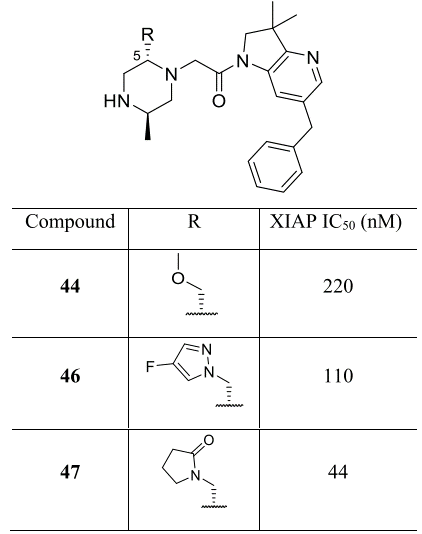
分子内ESP分析的第二个例子可以在IAP拮抗剂的进一步优化中找到,之前在蛋白质-配体相互作用的背景下讨论过58。从带有甲氧基甲基的化合物44(表3)开始,探索了哌嗪C-5取代基的SAR。在这一轮的优化过程中,注意到几个类似物的蛋白质-配体X-衍射共晶结构表明,新的取代基优先采用折叠构象,封填在中央酰胺键的顶部,如图16A所示的一个氮杂异构体母核子系列化合物45。与前面的那个例子一样,在识别出分子内堆积相互作用之后接着进行了溶液NMR研究,在本例中,化合物45的最低能游离构象与生物活性构象一致,如图16B所示,观察到了折叠的空间排列方式 。
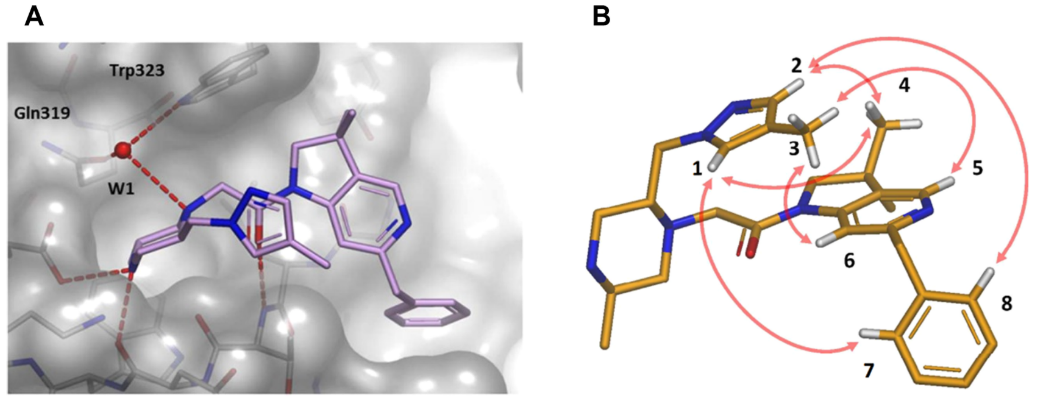
图16.(A)化合物45与XIAP-BIR3结合时X-衍射晶体结构。氢键显示为虚线。 结构化的水分子高亮显示为W1。(B)NMR观察到的化合物45溶液构象。箭头表示在NMR光谱中观察到的空间ROESY相关性。图片来源于J. Med. Chem. 2017, 60(11):4611−4625,经Taminini等人许可转载,版权归美国化学学会所有。
为了更好地理解这种相互作用并促进新化合物的设计以使得观察到的堆积方向最大化,于是计算了哌嗪-氮杂二氢吲哚母核和潜在的P2取代基的ESP表面(图17)。分析表明,许多基团的静电特征能与母核酰胺键形成最佳互补,这增强了偶极-偶极相互吸引的作用并有利于形成所需折叠式几何形状。图17显示了两个这样的例子:一个为芳香族,另一个为饱和杂环。
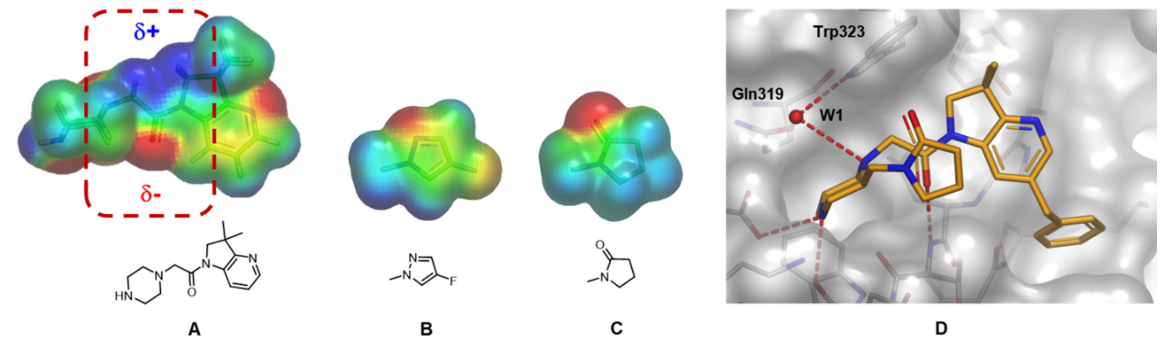
图17. 母核哌嗪-氮杂二氢吲哚骨架的ESP表面 (A) 和两个选出用于合成的哌嗪环上取代基示例 (B, C)。负值区域为红色,正值区域为蓝色。A酰胺键连接臂周围的关键区域用红色虚线高亮显示。(D) 化合物47与XIAP-BIR3的X-衍射共晶结构。氢键显示为虚线, 结构化的水分子突出显示为W1。图片来源于J. Med. Chem. 2017, 60(11):4611−4625,经Taminini等人许可转载,版权归美国化学学会所有。
对氟吡唑类化合物46和吡咯烷酮类化合物47的合成和生物活性测试表明,与母体化合物44相比(表3),化合物46与47的活性分别增强了2倍和5倍,其中化合物47在第一轮SAR探索中是配体效率最高的化合物(LE = 0.29)。与前面的示例一样,这种中等程度亲和力的增加伴随着LLE从3.1(化合物 44)到4.3(化合物 47)的改善,这表明细致的ESP分析可用于改善更多的理化性质。正如ESP模型所预期的那样,如图17所示,与XIAP-BIR3结合的化合物47的共晶结构证实了折叠构象的持久性。这个例子表明,即使在结合构象和游离构象最初相互一致的情况下,ESP分析也可用于分析分子内相互作用(在本例中为偶极-偶极堆积),并有助于设计有利于最大程度摆出生物活性构象的新目标分子。
5. 结论
从上面的例子可以看出,基于结构的药物发现项目可从ESP分析中获益并得以加速。已经证明改善静电互补性不仅可以改善化合物对蛋白靶标的亲和力,而且如CENP-E抑制剂算例所述的那样,还可以用来识别物理化学性质得以改善的可选骨架,从而提高细胞活性并改进体内特性。FXa因子抑制剂算例清楚地表明,在一个特定的系列中,需要考虑ESP的特性并加以细致地优化可以产生显著的活性提高。这种对芳香基团电子特性的优化让人想起了Topliss提出的用于优化芳香基团上取代基的决策树;然而,由于ESP可以预先计算,这将减少需要合成的化合物的数量,为给定蛋白靶标给出最有效的取代基组合。
ESP计算还提供了识别配体和蛋白之间非经典相互作用的机会,如IAP抑制剂算例所示。在骨架羰基上堆积芳香环可能不是开发有益相互作用的明显选择,但通过了解蛋白质和配体的ESP可以成功优化这种相互作用。使用ESP将CatS特异性抑制剂转化为CatS/CatL双重抑制剂的算例表明,通过巧妙地使用EPS表面,可以在针对一个靶标优化活性的同时而不会显着降低针对另一个靶标的活性。同样,虽然这里没有算例,但也可以想象一种情况,即ESP可用于设计针对特定反靶的选择性。
在配体ESP建模的两个算例(MDM2和IAP抑制剂)中,还进行了溶液构象研究,以进一步了解分子内相互作用的性质及其对蛋白质结合的能量学影响。然而,可以想象的是,即使在没有这种实验信息的情况下,单独的ESP分析仍然有助于化合物的设计并得出类似的结论。特别是在MDM2拮抗剂算例中,通过ESP计算可识别出两个芳环界面处的静电冲突,在任何情况下需要进一步探索分子这个位置的SAR,以及在随后更有利的类似物设计时并不特别依赖于溶液构象的知识。
结合溶液核磁共振实验或其他建模技术,从结构信息中识别或确认不利的冲突和所需相互作用的能力使分子内ESP分析成为SBDD中一项极具价值的技术。
从所讨论的算例还可以清楚地看出,虽然优化静电互补性的好处是显而易见的,但是药物化学课题组对该项技术的吸收有限。这种技术吸收受限一个原因可能是,从历史上看,即使对于小分子,产生ESP也是一种计算成本相对较高的技术,需要执行计算方面的专业知识。考虑到为蛋白质等大分子生成ESP的复杂性,这种计算成本呈指数级增长。此外,在真空中计算的带电分子的ESP表面实际上没有什么用处,可以通过隐式溶剂化模型进一步改进;然而,这进一步使程序复杂化,并增加了许多倍的计算时间。用于计算处理的图形处理单元 (GPU) 的兴起可能有助于降低了此类计算的障碍[60,61],但除了装备精良的实验室之外,它们可能仍然遥不可及。最近开发的用于生成近似DFT质量ESP 表面的GC-DNN模型比传统的QM方法快了许多个数量级(例如,一个典型的分子量为400的类药分子可能需要大约30分钟来计算DFT ESP;使用最近开发的GC-DNN模型可以将计算时间缩短到0.3秒),使用常规的计算机硬件,可以向历史上无法访问常规生成的QM ESP表面的实验室开放ESP生成。我们希望降低访问ESP数据的活化能垒,并成为增加ESP表面在药物化学中使用的催化剂。
6. 相关主题
- 用静电互补性快速、高效地优化配体-蛋白复合物的结合与选择性. http://blog.molcalx.com.cn/2019/04/04/flare-ec.html
- 如何用静电互补性打分. http://blog.molcalx.com.cn/2019/05/01/how-to-use-ec-score.html
- Cresset的核心技术. http://blog.molcalx.com.cn/2019/10/08/cresset-science-xed.html
- 探索更好的电荷模型. http://blog.molcalx.com.cn/2022/05/13/charge-model.html
- 更多的关于XED力场用于静电互补性分析算例:http://blog.molcalx.com.cn/tag/ec
- 最新版Flare V6不仅支持XED计算分子ESP表面,而且支持用QM计算分子EPS表面,参见:Flare V6预告——计算化学先睹为快
本文比较了DNN-ESP表面与XED力场计算ESP表面,结果表明两者非常相似。但是XED力场表面在计算带电分子上更具有优势,而DNN-ESP对带电分子计算EPS难以进行分析。因此强烈向你推荐用我们的Flare进行静电互补性分析。
7. 相关会议
- 2022年6月16日BST时间11:15,Astex的Gianni Chessari将在Cresset主办的Andy Vinter纪念会上做同名报告:Electrostatic Complementarity in Structure-Based Drug Design


