
摘要:本文介绍了两种偏心电荷模型:XED电荷模型与Astex-DNN电荷模型。XED电荷模型基于化学直觉、根据实验数据比较以改进电荷参数来开发的,是一种启发式模型;Astex-DNN电荷模型是用深度人工神经网络训练而来,是一种统计模型。本文对这两种电荷模型进行了比较,讨论了两者在电荷模型上的异同点以及在虚拟筛选性能上的比较,并提出了未来的开发方向。
作者:Peter Cherry/2021-08-04
编译:肖高铿/2021-08-10
蛋白-配体结合亲和力是预测药物活性最有用的指标之一。预估结合亲和力的技术正在不断发展变化,但有一件事保持不变——非共价相互作用的重要性。如何最好地预测这些相互作用是理论生化研究中最热门和最具挑战性的领域之一,几乎每周都会开发出新的方法,从完全经验的方法(例如1)到几乎完全从头计算的方法(例如2)。过去一年机器学习技术的兴起已经改变了药物发现的许多领域,非共价相互作用的预测也不例外。下面我们将介绍该领域最有前途的方法之一,将其与 Cresset的现有方法进行比较,并讨论这对未来Cresset软件开发意味着什么。
静电互补性(Electrostatic Complementarity™)
在深入讨论这些新进展之前,有必要花点时间了解一下静电互补性(Electrostatic Complementarity™,EC) 3的概念:它是什么、如何计算以及为什么它与药物设计相关。配体的EC是衡量配体的静电分布与您希望它结合的口袋的静电分布匹配程度的量度。如果EC很高,那么配体的静电势 (electrostatic potential,ESP) 表面为正的地方,在相应的蛋白质ESP表面都会有负电荷出现,反之亦然。换句话说,如果EC很高,那么蛋白质和配体之间的非共价相互作用在能量上是有利的,虽然EC不一定能直接预测结合亲和力,但是我们发现了两者之间具有非常好的相关性,特别是在同系物配体中,EC打分值和活性之间。
为了确定配体的EC,我们通常需要首先确定配体和蛋白质的ESP表面,这反过来又需要我们知道配体/蛋白质的电荷分布。不幸的是,从头算法预测电荷分布,例如,通过使用来自后Hartree-Fock计算的电子密度,计算量太大,以至于它实际上对虚拟筛选没有什么用。因此,通常采用近似电荷模型。 Cresset使用自己的XED力场4,它已被证明是高效、准确的5。然而科学并没有停滞不前,在本文中,我们问自己:XED是否仍然是最佳选择。
已经有许多令人眼花缭乱的替代方法,从RESP6等全电子密度方法、AM1-BCC7等原子电荷模型、到AMOEBA8等多极矩方法。也许最令人兴奋的方法之一是Astex制药9最近开发的电荷模型,此后称为Astex-DNN,它在许多方面与XED相似:比如通过将电荷分配给分子内的点、每个原子一个电荷、给分子中的“特征点”分配更多的电荷来定义分子的电荷分布。这些特征点包括:孤对电子、σ-孔、π-轨道等的近似位置。然而,Astex DNN与XED在一些关键特征上显著的不同。XED力场是通过化学直觉、与实验数据比较以改进电荷参数来开发的,而Astex-DNN电荷是使用经过训练的动态神经网络 (DNN) 确定的,经过训练该网络可以重现高级二阶Moller-Plesset计算的ESP表面的电荷。所以,本质上,我们有一个启发式模型(XED)和一个统计模型(Astex-DNN)。虽然启发式模型具有更易于理解的优势,但后者可能会在标准化学启发式无法解决的更多“外来”分子上胜出。此外,Astex-DNN的改进可能不需要对模型有任何的重大化学洞察或修改;只需扩展用于训练DNN的数据集并重新训练模型。
比较Astex-DNN与XED模型的性能
为了比较Astex-DNN和XED模型的性能,我们提出了四个主要问题:
- 两个模型预测的电荷有多相似?
- 生成的ESP表面有多相似?
- 这两种方法在配体叠合计算中的表现有多相似?
- 这两种方法的相对速度如何?
电荷点的相似性
Astex-DNN和XED模型将电荷分配给原子上的点,以及位于电子结构中重要位置的非原子点。虽然两个模型的原子点的位置必须相同,但非原子点的位置和数量不同。图1显示了XED和Astex-DNN的非原子点,它们的位置大致一样,但存在一些关键差异。XED模型的非原子点比 Astex-DNN模型的更靠近原子。此外,它们对电子结构的某些特征使用了不同数量的电荷点,例如,XED使用多个非原子电荷点来描述孤对电子,而Astex-DNN仅使用一个。
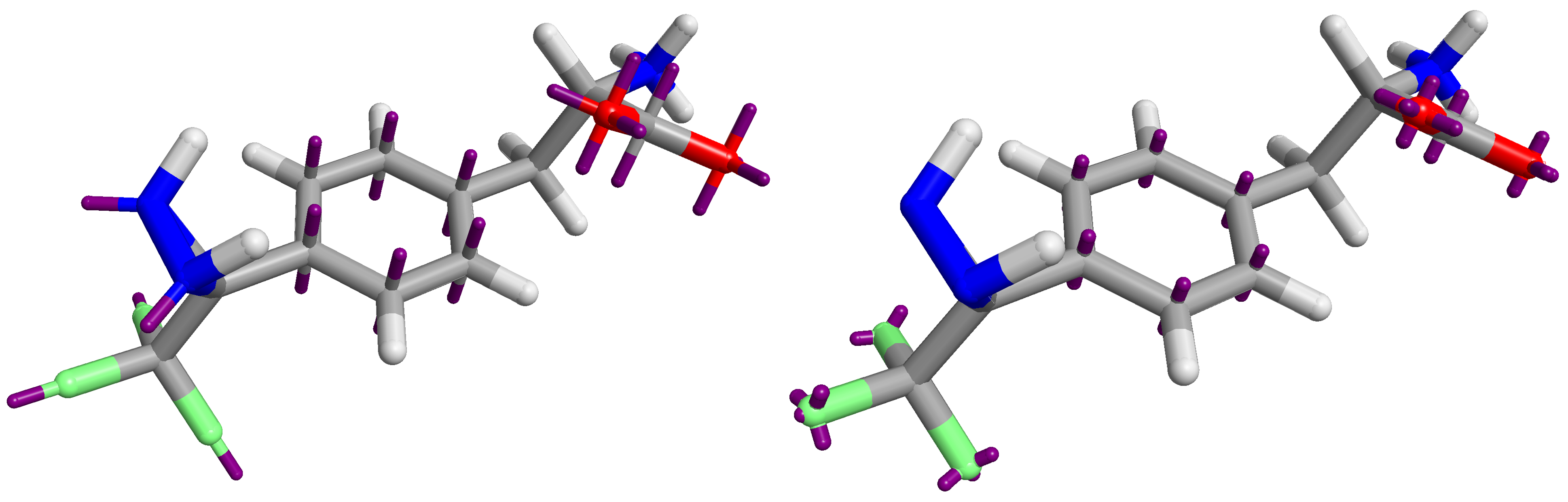
图1. 紫色线指示了非原子电荷点的位置。左:图1a(Astex-DNN),右:图1b (XED)。
非原子电荷点位置的差异使得分配给电荷点的电荷值的直接定量比较没有实际意义,但从静电势最大值的比较我们可以看到模型似乎非常相似(图 2)。
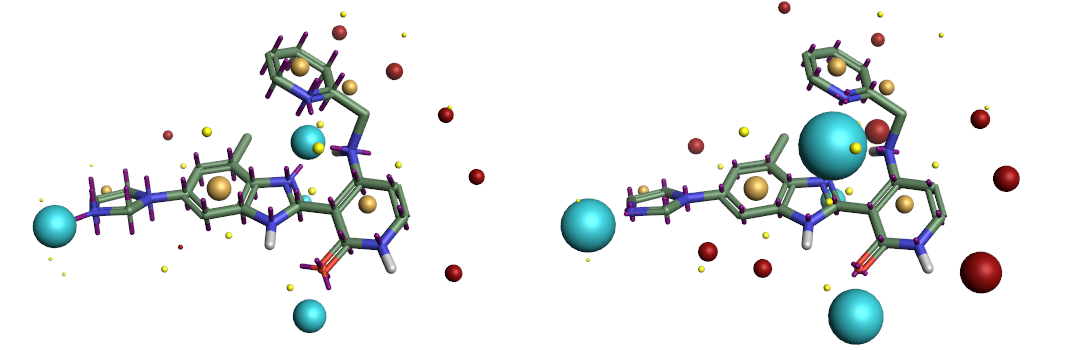
图2. 两种电荷模型计算得到的静电势,小园球为场点(Field point)指示了静电势极大、极小点。左:图2a(Astex-DNN),右:图2b (XED)。
ESP表面相似性
ESP表面的比较很有用,因为若差异存在则表明至少有一个模型是不正确的,并且可能模型存在缺陷。鉴于模型使用不同的非原子电荷点集合,预测的ESP表面几乎肯定会有所不同;唯一不会的情况是在罕见的对称情况下,或者当非原子点上的电荷为零时。然而,尽管存在这一关键差异,Astex-DNN确实预测了稍微更大程度的极化作用,但是我们发现预测的ESP表面惊人地相似。一个典型的例子如图3a和3b所示。
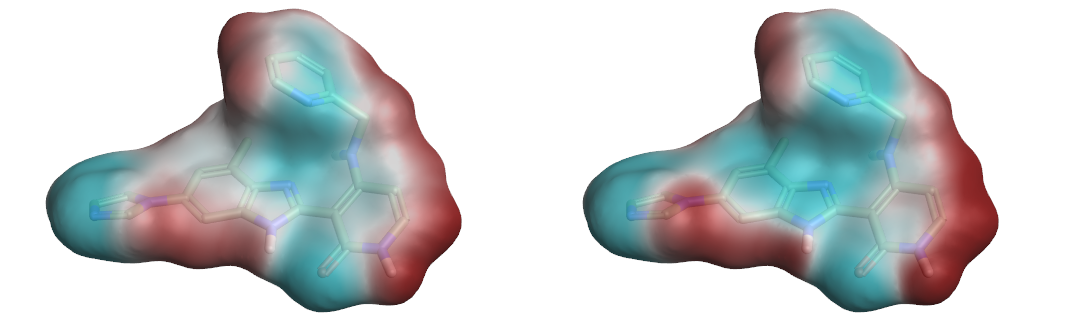
图3. 两种不同电荷模型计算的ESP表面。左:图3a(Astex-DNN),右:图3b (XED)。
配体叠合的性能
由于此类计算在虚拟筛选中起着至关重要的作用,因此一个令人感兴趣的测试是查看两个模型在配体叠合计算中的性能表现如何。该测试回答以下问题:如果我们知道两个配体A和B的静电特征,它们都已知与一个蛋白结合,我们是否可以通过一个配体的生物活性构象来确定另一个配体的生物活性构象?
为了回答这个问题,我们需要两个实验结构:一个是蛋白与配体A的复合物结合,另一个是同一个蛋白与配体B的复合物结合。第一步是将这两个复合物结构进行叠合。这将是我们知晓配体A的生物活性构象与配体B的生物活性构象是如何叠合的,我们称之为“正确”的叠合。
生成配体B的构象异构体,然后尝试将配体B的每一个构象异构体叠合到配体A的生物活性构象上。至关重要的是,这种叠合不是基于两种构象的原子结构,而是仅仅依赖于静电特征的相似性。因此,与配体A生物活性构象叠合最佳的配体B的构象将是静电分布具有最大重叠的构象。然后,通过比较静电特征获得配体B的“最佳”构象,并与通过比较蛋白-配体复合物结构获得的配体B的“正确”构象进行比较。通过计算配体B的这两种构象的原子位置的均方根差(RMSD)来进行结构比较。如果RMSD较低,则表明静电相似性是识别配体生物活性构象的有用工具。由于静电相似性由静电分布确定,而静电分布又由电荷模型确定,因此当试图识别配体的生物活性构象时,该测试将可回答两个电荷模型中哪一个更好的问题。
在理想的情况下,配体B的这两种结构是相同的,但实际上情况并非如此。第一个原因是配体的静电分布并不是确定构象的唯一因素,并且即使原子结构相当大的变化对静电分布的影响相对较小。 第二,我们对构象空间的采样不可避免地是不完整的,因此配体B的生物活性构象可能不在我们产生的构象集合中。第三,不能保证配体A和配体B具有相同的结合模式,即使它们结合到相同的口袋。 如果是这种情况,那么具有与A的生物活性构象最相似的静电分布的B的构象可能不是结合在蛋白口袋中时所采取的构象。尽管有这些缺点,但这是一个非常有用的测试,因为它直接与这些电荷模型的实际使用相平行。
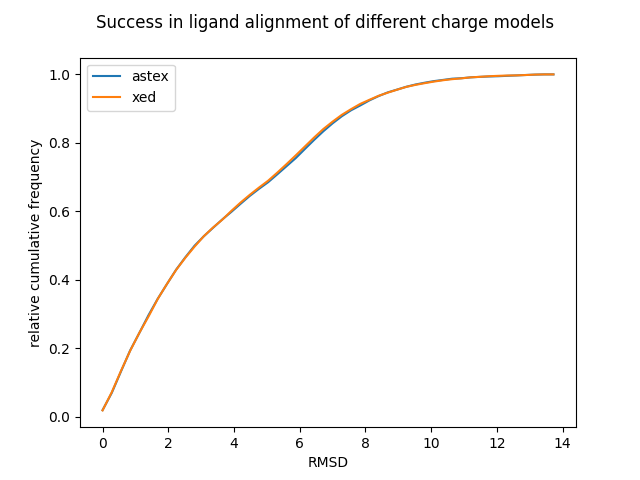
图4. 相对累积频次图,表明了两种电荷模型在配体叠合计算中的性能。左:图1a(Astex-DNN),右:图1b (XED)。
图4显示了Astex-DNN和XED电荷模型的生物活性构象和具有最高EC打分值构象之间RMSD的累积频次图,低RMSD的构象越多越好。该测试是在从阿斯利康/剑桥晶体数据中心10获得的119个蛋白与约1450个配体上进行的。
这两个模型的性能非常相似,令人不安。但是,结果中存在许多明显的差异,这被非常大的数据集所掩盖。 两种模型都不是100%准确的, 这是可以预料的,生物活性构象在几何形状上的微小变化不会对配体-蛋白结合强度产生显著影响。此外,值得注意的是,在对结果后进行更仔细的检查时发现两个电荷模型具有相似的失效算例,即,如果一种电荷模型预测了错误的构象,那么另一个电荷模型也可能会预测错误。
结论
所进行的测试表明,XED和Astex-DNN电荷模型表现出非常相似的性能,并且任意一个都是虚拟筛选计算的良好选择。在某些方面,这令人失望,因为它表明:尽管机器学习技术取得了巨大进步,但是复杂的神经网络仍然不能优于基于人类直觉的启发式算法。另一方面,它非常令人放心,因为它表明用于构建原始XED力场的直觉和概念是正确的。
两种模型之间缺乏明确的区别绝不代表电荷模型的发展陷入僵局。首先,机器学习技术在药物发现中的应用目前还处于初级阶段,我们可以期待更多令人兴奋的发展。事实上,开发一种能够与XED力场的性能相当的模型,而不依赖于化学理论的启发式,是一项了不起的成就。通过简单扩展用于训练模型的数据集,就有可能获得一个性能优于XED 的模型。然而,还有一个更让人感兴趣的发展路径可以讨论。
将来的工作:电荷体系
XED和Astex-DNN都对带电配体性能表现不佳。造成这种情况的一个关键原因是,如果一个分子具有带电原子,那么从真空DFT计算获得的 ESP表面将完全被该电荷主导。这不是DFT计算中的错误;它是ESP表面的精确表示,但这种单调的ESP表面区分特征缺乏意味着配体叠合计算难以精确执行。从本质上讲,大小和形状相似的带电配体的ESP表面最终看起来都差不多,从而阻止了它用来识别哪些配体会与蛋白结合,哪些不会。幸运的是,这个问题的根源很清楚。用于生成Astex模型的训练数据的DFT计算是在真空中进行的;我们真正感兴趣的是:当配体在结合口袋中时,配体的ESP表面是什么。配体与口袋之间的相互作用可导致形式电荷更加集中在分子内的特定基团上,并配体上的总电荷减少。因此,如果我们要对带电配体进行配体叠合计算,我们需要知道这些配体-口袋相互作用是什么,以及它们如何影响配体的ESP。
不幸的是,这种相互作用的性质难以预测。虽然我们可以假定:如果配体上有一个带电荷基团,那么在口袋内的互补点上肯定有一个带相反电荷的基团,目前尚不清楚如何确定该点上的电荷强度以便正确表示蛋白-配体相互作用。从多个方面讲,这是使用ESP表面来描述蛋白-配体结合的缺点。虽然它是衡量两者之间相互作用强度的一个好指标,但它并非完整,并且有一些重要的过程(例如氢键)取决于两种成分的化学组成。
目前,ASTeX-DNN和XED模型都使用粗略的方法来解决这个问题。 两者基本上都依赖于将电荷定位于官能团上并将电荷的大小用一个因子按比例缩小以确保该带电基团不会完全支配ESP分布。考虑到这些方法的特殊性质,它们的性能表现得非常好,但带电荷配体的叠合结果仍然比中性配体更具挑战性。改善这一点是未来的一个关键挑战,也是Cresset正在进行的研究主题。
参考文献
- Colwell L.J., Statistical and machine learning approaches to predicting protein–ligand interactions, Current Opinion in Structural Biology 49, 123-128 (2018) doi:10.1016/j.sbi.2018.01.006
- Gundelach L., Fox T., Tautermann C.S., & Skylaris C.-K., Protein–ligand free energies of binding from full-protein DFT calculations: convergence and choice of exchange–correlation functional doi:10.F1039/D1CP00206
- Bauer M, Mackey MD, Electrostatic Complementarity as a Fast and Effective Tool to Optimize Binding and Selectivity of Protein–Ligand Complexes https://pubs.acs.org/doi/abs/10.1021/acs.jmedchem.8b01925
- Molecular Field Extrema as Descriptors of Biological Activity: Definition and Validation. J. Chem. Inf. Model. 2006, 46 (2), 665-676 https://pubs.acs.org/doi/abs/10.1021/ci050357s
- Chessari G., Christopher A. Hunter C.A.., Low C.M.R., Packer M.J.., Vinter J.G., Zonta C. An Evaluation of Force-Field Treatments of Aromatic Interactions, An Evaluation of Force-Field Treatments of Aromatic Interactions, (2002) doi:10.1002/1521-3765(20020703)8:13<2860::AID-CHEM2860>3.0.CO;2-N
- Bayly, C.I., Cieplak, P., Cornell, W., & Kollman, P.A., A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model. The Journal of Physical Chemistry 97, 40 (1993) doi: 10.1021/j100142a004
- Jakalian, A., Jack, D.B., & Bayly, C.I. (2002). Fast, efficient generation of high‐quality atomic charges. AM1‐BCC model: II. Parameterization and validation. Journal of computational chemistry 23, 16, (2002) doi: 10.1002/jcc.10128
- Rackers, J. A., Wang, Q., Liu, C., Piquemal, J.P., Ren, P., & Ponder, J.W., An optimized charge penetration model for use with the AMOEBA force field. Physical Chemistry Chemical Physics 19, 1, (2017) doi: 10.1039/C6CP06017J
- Rathi, P.C., Ludlow, R.F., & Verdonk, M.L. (2019). Practical high-quality electrostatic potential surfaces for drug discovery using a graph-convolutional deep neural network. Journal of medicinal chemistry 63, 16, doi: 10.1021/acs.jmedchem.9b01129
- Giangreco, I., Cosgrove, D. A., Packer M. J., (2013) An Extensive and Diverse Set of Molecular Overlays for the Validation of Pharmacophore Programs, Journal of Chemical Information and Modelling, 53, 4, doi: 10.1021/ci400020a
推荐阅读
- Bauer, M. R.; Mackey, M. D. Electrostatic Complementarity as a Fast and Effective Tool to Optimize Binding and Selectivity of Protein–Ligand Complexes. J. Med. Chem. 2019, 62 (6), 3036–3050. https://doi.org/10.1021/acs.jmedchem.8b01925.
- Cons, B. D.; Twigg, D. G.; Kumar, R.; Chessari, G. Electrostatic Complementarity in Structure-Based Drug Design. J. Med. Chem. 2022. https://doi.org/10.1021/acs.jmedchem.2c00164.
- 如何用静电互补性打分. 墨灵格的博客. http://blog.molcalx.com.cn/2019/05/01/how-to-use-ec-score.html


