
用水的能量学信息重现KRASG12D选择性抑制剂MRTX1133的哌嗪侧链优化过程
摘要:本文演示了如何用SZMAP/GAMEPLAN对KRAS G12D-化合物5B的共晶结构(PDB 7RT4)结合位点进行水能量学分析,生成对5B进行结构优化的取代假设。结果表明,Gameplan给出了正确的C7位萘环修饰建议,更重要的是,给出了两种可能的C4位哌嗪环修饰建议:大环化取代假设与桥环取代假设。其中大环化修饰假设已经被AZD4625所证实,哌嗪环桥环化修饰假设也与现有结果一致。将哌嗪环与vdw取代假设作为query的侧链虚拟筛选,也在打分最高的化合物里命中高活性的桥环化哌嗪。总的来说,SZMAP/GAMEPLAN根据水能量学分析的取代假设重现了MRTX1133关键侧链优化的设计,可用于基于结构设计为药化提供直观的思路。
前言
MRTX1133(图1,化合物1)是Mirati Therapeutics公司开发的靶向KRASG12D选择性的强效、非共价抑制剂,目前处于临床前阶段,现有数据显示出巨大的进一步开发潜力[1]。
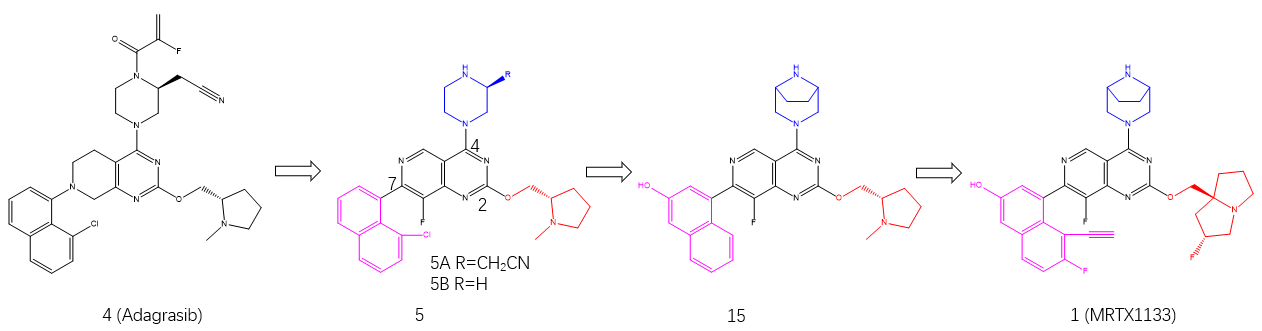
图1. MRTX1133的发现过程
MRTX1133的发现过程关键步骤如图1所示[1]。根据在发现KRASG12C抑制剂adagrasib(化合物 4)方面的经验,作者合成了吡啶骈[4,3-d]嘧啶骨架化合物5A和5B,并预期质子化的哌嗪可以与突变体的ASP12侧链相互作用而有效。尽管化合物5A没有活性,但不含氰甲基的类似物5B在加入GDP的KRASG12D的SPR测定中显示KD=3.5μM的结合亲和力。作者决定以5B为起点对吡啶骈[4,3-d]嘧啶骨架化合物进一步进行优化。5B与KRASG12D的共晶结构(PDB 7RT4)表明,B15结合到KRASG12D的开关II口袋(图2),类似于adagrasib与KRASG12C的结合模式。
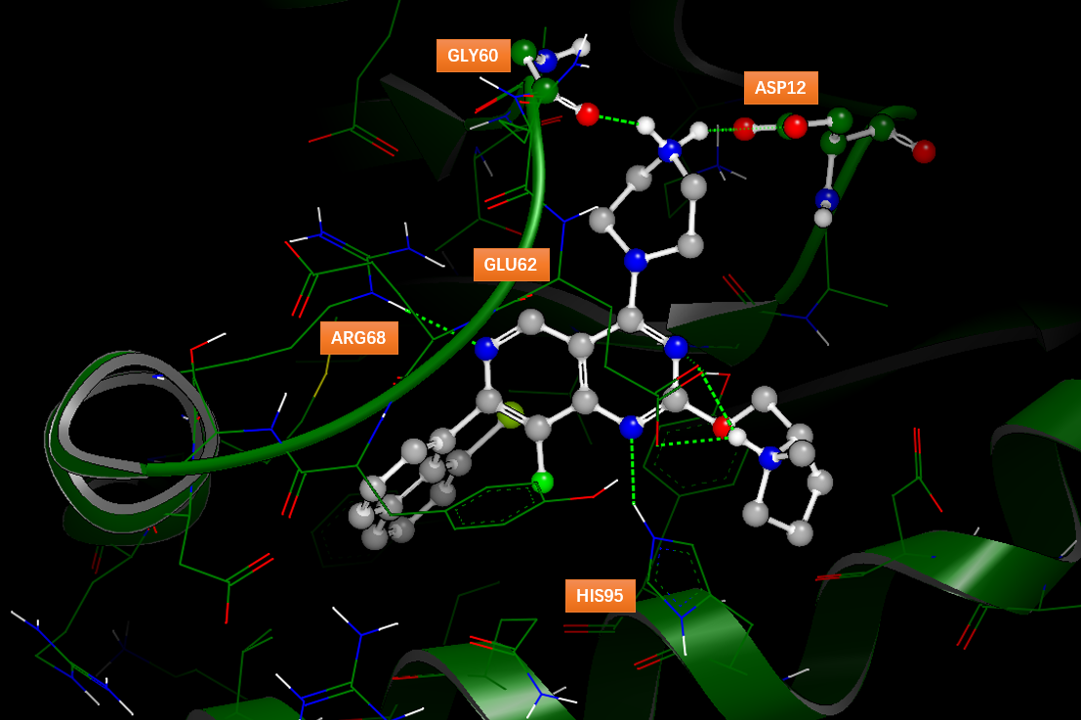
图2. 化合物B5与KRASG12D的结合模式(PDB 7RT4)
如图2所示,5B的6-位氮与Arg68发生氢键相互作用,并且8-位氟占据在一个小的疏水口袋中,这可能解释为什么吡啶并[4,3-d]嘧啶母核比adagrasib相应的四氢吡啶并嘧啶更有效(后者结合亲和力降低了5-10倍)。吡啶并[4,3-d]嘧啶母核的2、4、7-位(见图1 5B)上不同的取代其分别与口袋不同位置发生相互作用。C4-位的质子化哌嗪与突变体ASP12形成离子对盐桥相互作用,这使得对KRASG12D比KRAS WT高10倍的选择性。哌嗪环采取了一个高能的扭船式构象,这与之前在Adagrasib-KRASG12C复合物结构中观察到的一样(PDB 6UT0),哌嗪正电中心与Asp12的盐桥以及与GLY60羰基氧的氢键对高能扭船式构象起到了稳定作用。C2-位取代基处的保守水分子(未在图3呈现)与GLY10和THR58形成氢键,但与化合物5B没有明显的相互作用,这为进一步的探索提供了机会。C2-位带正电荷的吡咯烷基片段非常适合与GLU62发生有利的盐桥相互作用。最后,C7-位萘基占据了一个深的疏水口袋,正如先前报道的adagrasib一样。总的来说,2、4、7这三个位置上的取代基被确定为增加对G12D蛋白亲和力的机会。
表1. 吡啶骈[4,3-d]嘧啶母核4-位的探索
![吡啶骈[4,3-d]嘧啶母核4-位的探索](http://blog.molcalx.com.cn/wp-content/uploads/2022/05/2022051514232878.png)
鉴于与Asp12的盐桥对5B先导化合物结合和选择性的重要性,作者使用了在KRASG12C工作中确定的C7-萘酚为基础来对C4位进行优化,部分结果如表1所示。未取代哌嗪基化合物6的亲和力为0.19μM,而高哌嗪类似物7的亲和力低了2倍,N-甲基化的8以及用氧取代仲氨基的9都是对活性不利的。此外,化合物8和9失去了对KRAS WT 的选择性(SPR KD分别为6.2和0.56μM)。这些结果进一步证实了质子化哌嗪与Asp12及Gly60的相互作用对活性和选择性是非常重要性的。甲基取代的哌嗪10和11并未提高活性。然而,生物活性构象刚性的哌嗪衍生物12-15却增加了亲和力。具有[3.2.1]双环二氨基取代的化合物15对KRAS WT的结合亲和力KD=182nM,而对KRASG12D的KD=0.8nM,选择性超过200倍。与增加的亲和力一致,15也观察到明显的细胞活性(针对 AGS,KRASG12D细胞系的pERK IC50=0.530 μM)。这为识别有效的KRAS G12D抑制剂的提供了希望。

图3. 化合物15与KRASG12D的结合模式(PDB 7RT1)
化合物15与KRASG12D/GDP的X-射线衍射结构(PDB 7RT1)如图3所示。双环基团的双碳桥占据了一个小口袋,一个内式C-H与GLY10羰基氧形成非经典氢键相互作用,这有助于将带电荷的仲胺固定在位置上以实现与Asp12和Gly60的最佳相互作用。鉴于此,作者使用基于化合物15的探针开发了KRASG12D/GDP的HTRF测定方法以提高筛选通量,HTRF测定的相对活性排序与SPR亲和力可以很好地对齐(表 1)。
鉴于从化合物5B到15的C4-位哌嗪优化(引入桥环活性提高283倍)是实现选择性与提高活性的关键步骤,本文的主要目的是探索能否借助基于结构的方法实现C4-位从哌嗪到桥环哌嗪的优化。之前我们[2]已经用SZMAP[3]分析过KRASG12C结合位点的水能量学特征,解释共价抑制剂MRTX849先导化合物优化过程中的SAR,其中包括对C4-位哌嗪环片段的构象、腈基的对水的替换关系以及C7-位萘环羟基引入对活性的影响。除了通过绘制等值图分析结合位点水能量学特征之外,还可以用SZMAP/GAMEPLAN直接对结合位点里配体给出结构优化与修饰的建议。因此,更具体的讲,本文的主要目的是用GAMEPLAN对5B与C4位哌嗪环进行分析,看等否得到桥环修饰的建议以重现该部分的优化过程。
方法与材料
5B与KRASG12D复合物结构从PDB上下载(PDB code:7RT4),用Spruce进行标准的结构准备。
1 | spruce -in 7rt4.pdb |
对准备好的结构(7RT4_AaltA__DU__7IZ_A-203.oedu)用SZMAP/GAMEPLAN进行分析,采用默认参数:
1 | gameplan -szmap_mpi_np 28 -p 7RT4_AaltA__DU__7IZ_A-203.oedu -prefix 7rt4_gameplan |
仅需1分钟左右即计算完毕,得到结果文件7rt4_gameplan.oeb.gz,用可视化软件Vida进行分析:
1 | vida 7rt4_gameplan.oeb.gz |
结果与讨论
5B的结构修饰假设
GamePlan对蛋白-配体结合位点进行分析以确定从溶剂信息可能令人感兴趣的点,对这些点进行SZMAP计算,然后处理结果,最后得到配体修饰的各种假设并识别对复合物起到稳定化或去稳定化作用的水位点。
首先查看vdw与nopolar取代的gameplan假设,如图4所示。
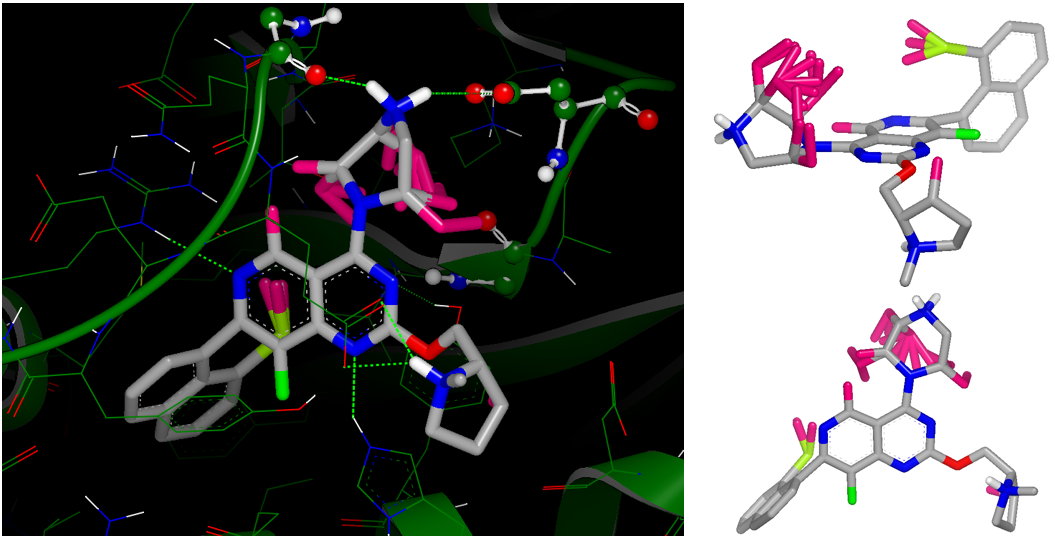
图4. vdw/nopolar取代的GAMEPLAN假设,其中右图为了清晰起见而给出两种不同的视角。
在图4中的粉红色部分是从配体延伸出2个键范围内引入vdw/nopolar取代的假设。可以看到,在C4位置哌嗪环上聚集了很多的引入新基团的机会,尤其是在哌嗪环面向Asp12的一侧上(见图4右上)。通过进一步对哌嗪部分该信息的分析与浏览,很容易得到对哌嗪环进行桥连的假设,如图5所示。
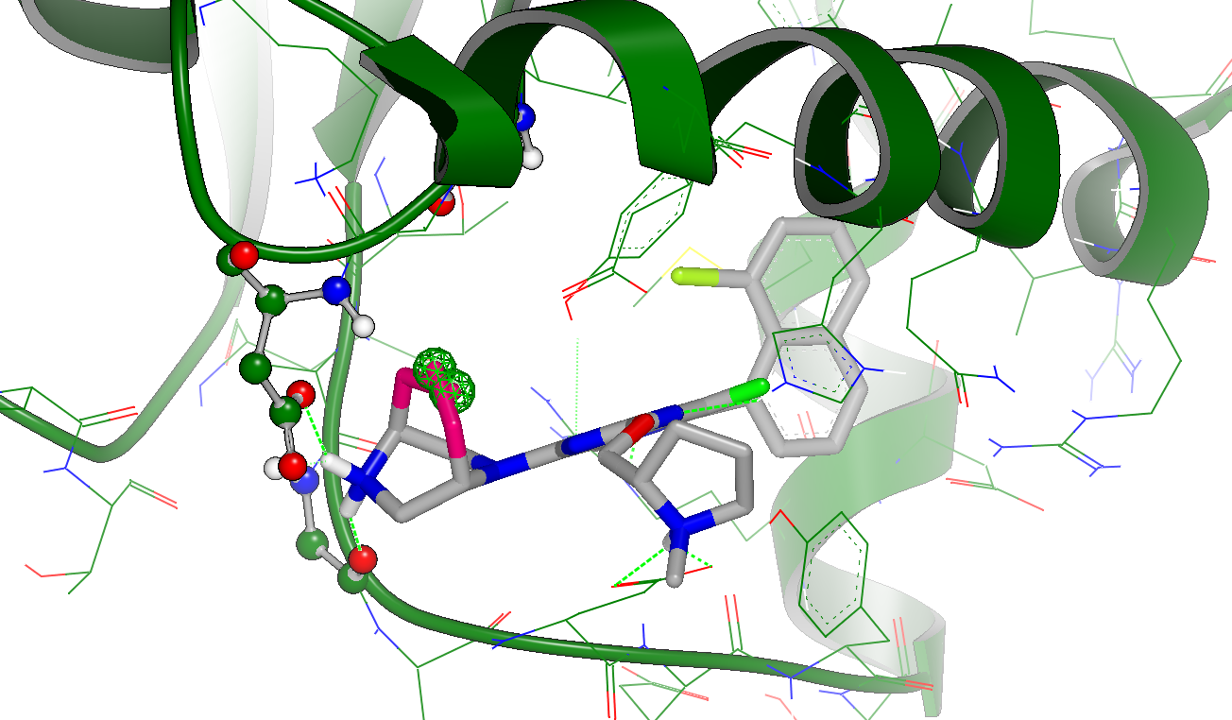
图5. C4位哌嗪环部分的vdw取代假设
Gameplan的假设还包括在母核与哌嗪环之间成环,如图6红色圆圈高亮显示的部分:在此提示下,足以让有经验的化学家设计出大环分子以稳定哌嗪环的构象。阿斯利康最近公开的KRASG12C共价抑制剂AZD4625[4]就属于此类设计,这也证明了gameplan的取代假设是正确。
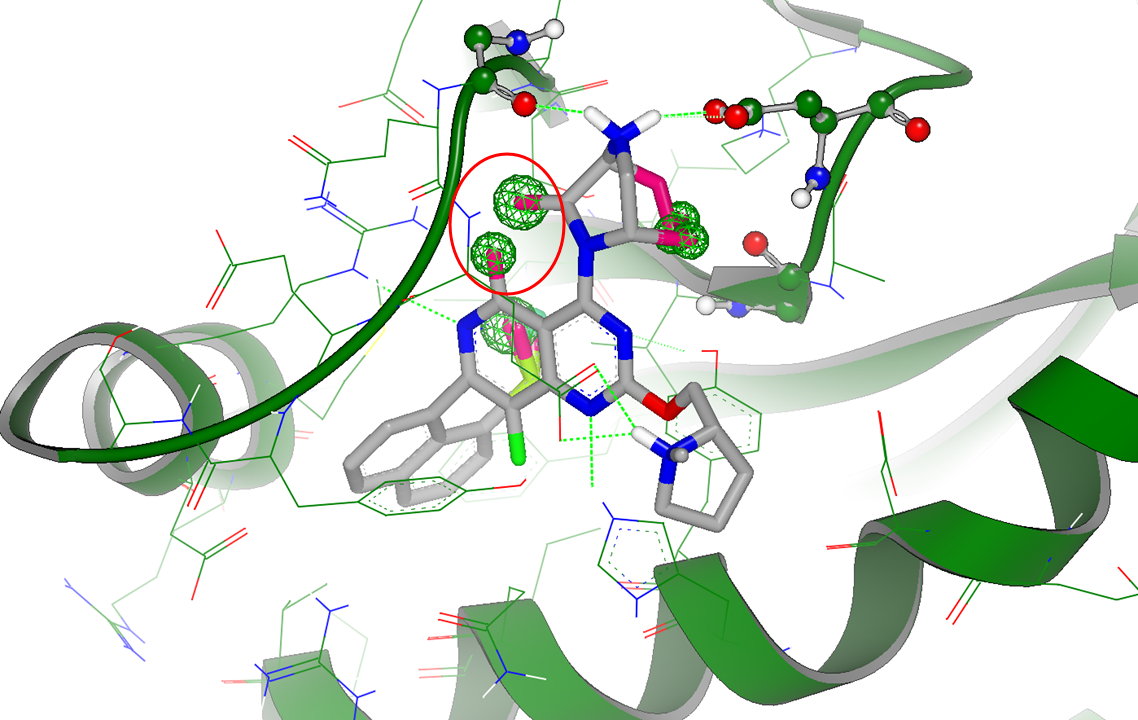
图6. 与C4位哌嗪环有关的大环vdw取代假设
虽然在C7-位萘环的氯原子上与C2-位四氢吡咯环上也有vdw取代机会,但不是本文的重点,因此不对这两个位置进行讨论。
现在来看一下极性取代基的机会,图7的粉红色棍状末端是gameplan生成的全部极性取代假设。
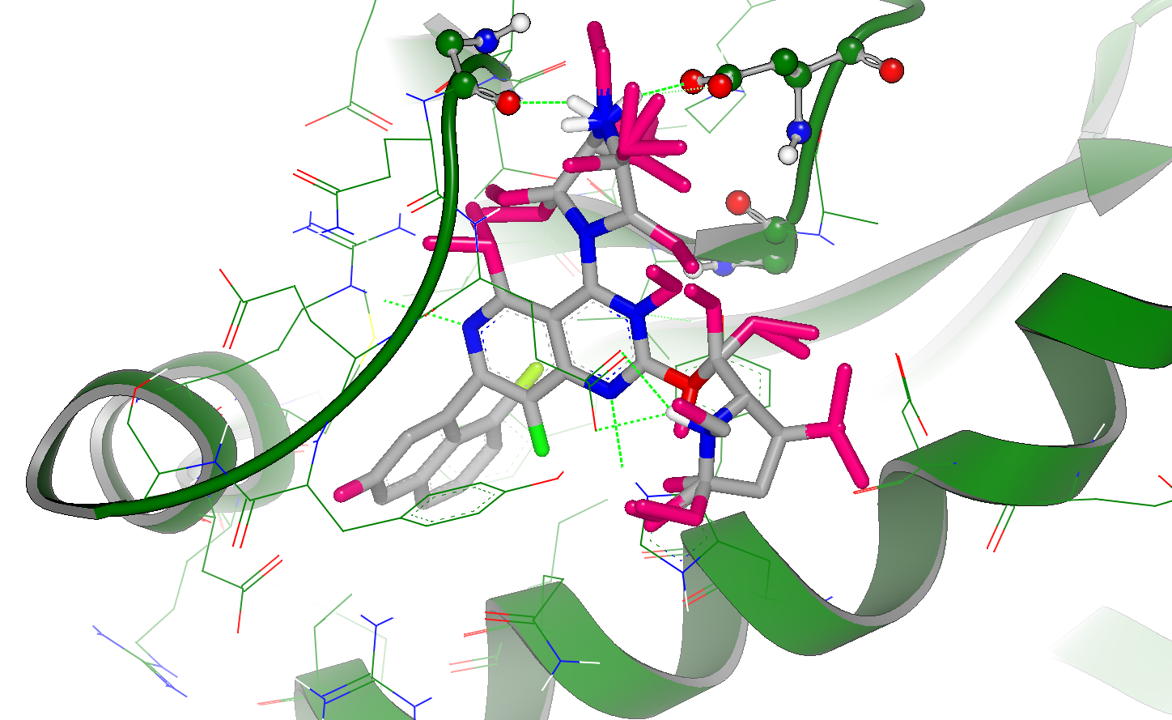
图7. gameplan生成的极性取代假设
如图7所示,注意到在萘环上有个极性基团取代机会,在C4位哌嗪环朝向Asp12方向之前vdw桥环取代假设、大环化取代假设的位置上也出现有极性取代,这提示可重点进一步对这些位置的极性取代进行分析,图8呈现了vdw与polar取代重合的位置。
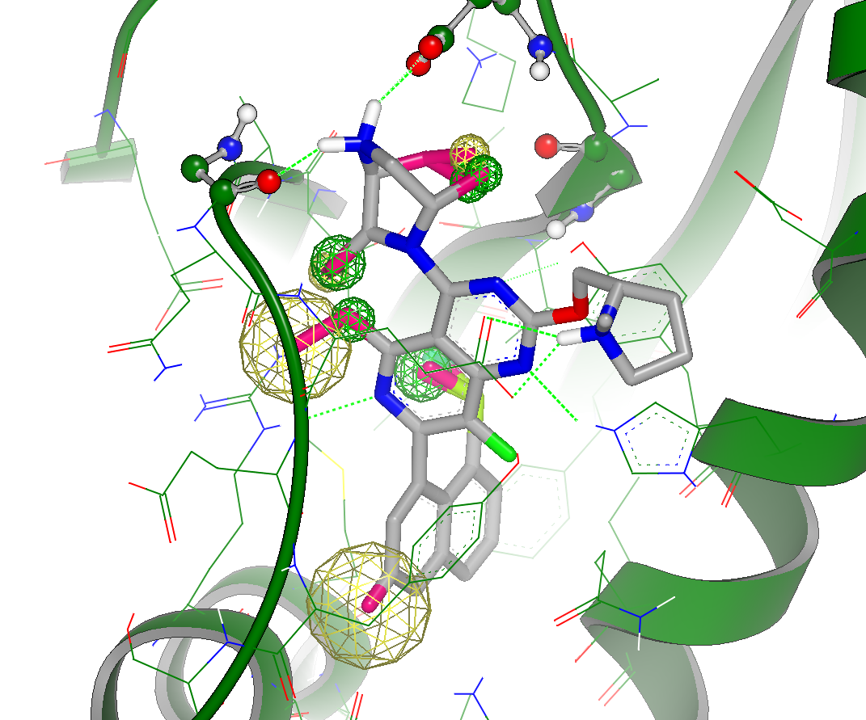
图8. 与哌嗪环相关的gameplan的极性取代假设以及萘环极性取代假设。黄色网格状球:极性取代;绿色网格状球:vdw取代,球的大小表示强度。
如图8所示,萘环部分有个大的黄色网格球,表示这里推荐用极性基团取代,之前在对MRTX849进行分析时也存在同样的极性基团取代建议[2],这与MRTX1133的萘环羟基对应。在vdw取代连接母核与哌嗪的假设中,在潜在的大环两侧存在黄色网格球,这在图9中隐去了蛋白结构之后的视图更加清晰地看到;在哌嗪环面向Asp12一侧的vdw取代基础也出现黄色网格球,这意味着该处引入的取代基可以是极性的,这与MRTX1133桥环内式的C-H作为非经典氢键供体是一致的。

图9. 与哌嗪环相关的gameplan的极性取代假设以及萘环极性取代假设——隐去蛋白结构的视图。黄色网格状球:极性取代;绿色网格状球:vdw取代,球的大小表示强度。
C4位侧链虚拟筛选
在上述根据水能量学而产生的gameplan取代假设基础上,对C4位的哌嗪部分我们可以构造一个被vdw取代的哌嗪query,用BROOD或SPARK对数据库进行虚拟筛选可发现满足条件的新片段来代替哌嗪。结果如图10所示,在打分靠前的化合物里包含了表1活性得以提高的部分化合物,其中包括MRTX1133的桥环哌嗪。这说明,szmap/gameplan取代假设结合片段虚拟筛选,可以实现对B5 C4位哌嗪片段进行高效优化目的。
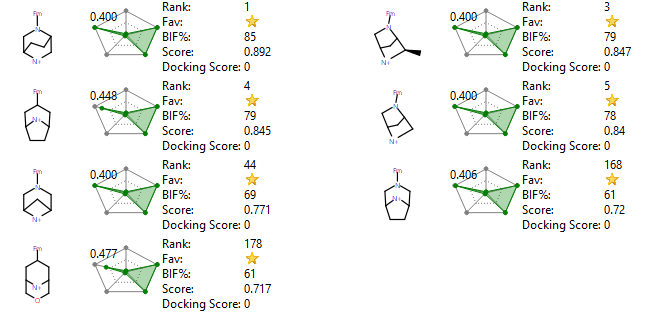
图10. 对C4位用vdw取代的哌嗪为模板进行片段虚拟筛选后部分打分靠前的片段
结论
本文演示了如何用SZMAP/GAMEPLAN对KRAS G12D——5B的共晶结构(PDB 7RT4)的结合位点进行水能量学分析,生成对5B进行结构优化的取代假设。结果表明,Gameplan不仅给出了正确的C7位萘环修饰建议,更重要的是,给出了两种可能的哌嗪环修饰建议:大环化取代假设与桥环取代假设。其中大环化修饰假设已经被AZD4625所证实,在哌嗪环面向Asp12羧基一侧引入桥环的取代也复现了现有高活性化合物结构。这说明,用简单的水能量学分析,可以直接指导C4位哌嗪环的优化。
将哌嗪环与vdw取代假设作为query进行侧链虚拟筛选,也在打分最高的化合物里命中最佳的桥环化哌嗪,这极大地加速了优化过程,直接在gameplan假设的基础上给出了目标化合物的建议。
总的来说,SZMAP/GAMEPLAN根据水能学分析的取代假设可以重现MRTX1133关键侧链优化的设计。
补充:关于AZD4625的大环化设计
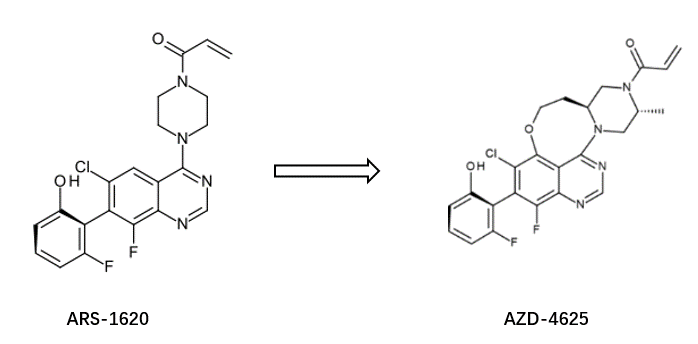
图1S. 化合物ARS-1609与AZD4625的化学结构
前面提到阿斯利康Kettle等人[4]最近公开的化合物AZD4625(见图1S 右)可以视为ARS-1620的环约束衍生物,旨在限制哌嗪环的构象。这个设计也可从SZMAP/Gameplan的水分子替换策略中获得提示。对ARS-1620与KRASG12C的共晶结构PDB 5V9U进行分析,可以获得如图2S的vdw与nopolar取代假设。
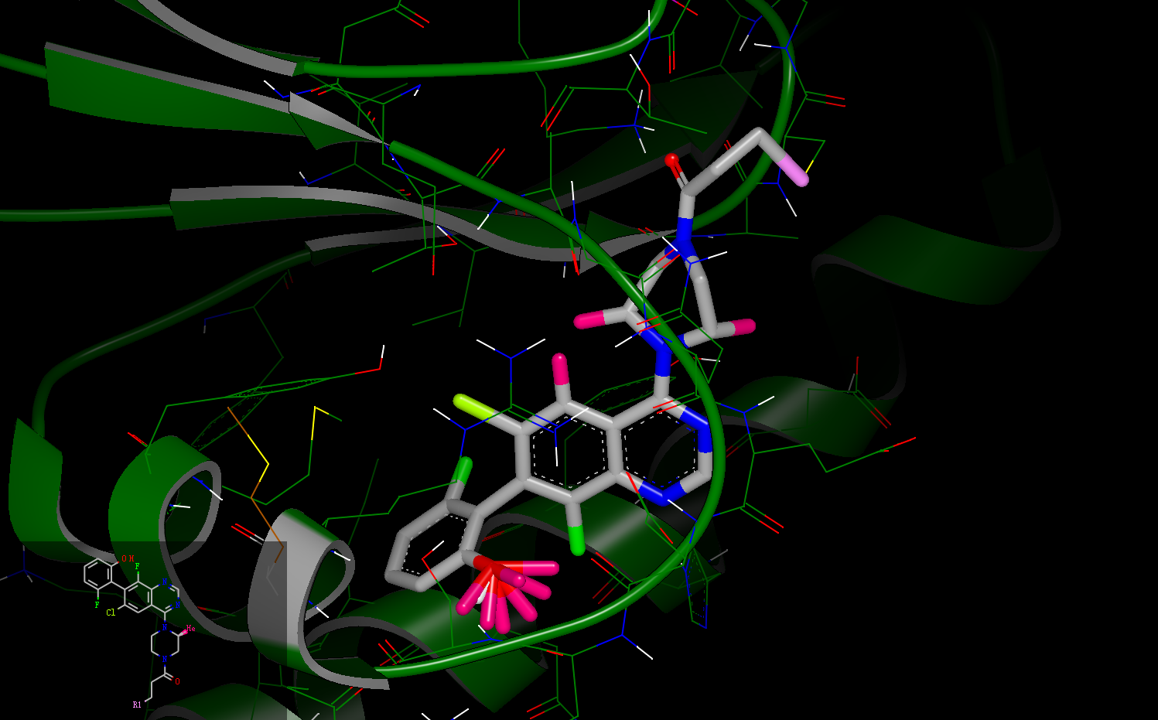
图2S. ARS-1620的vdw与非极性取代假设
SZMAP/gameplan还给出了可以如图3S所示的polar取代假设。
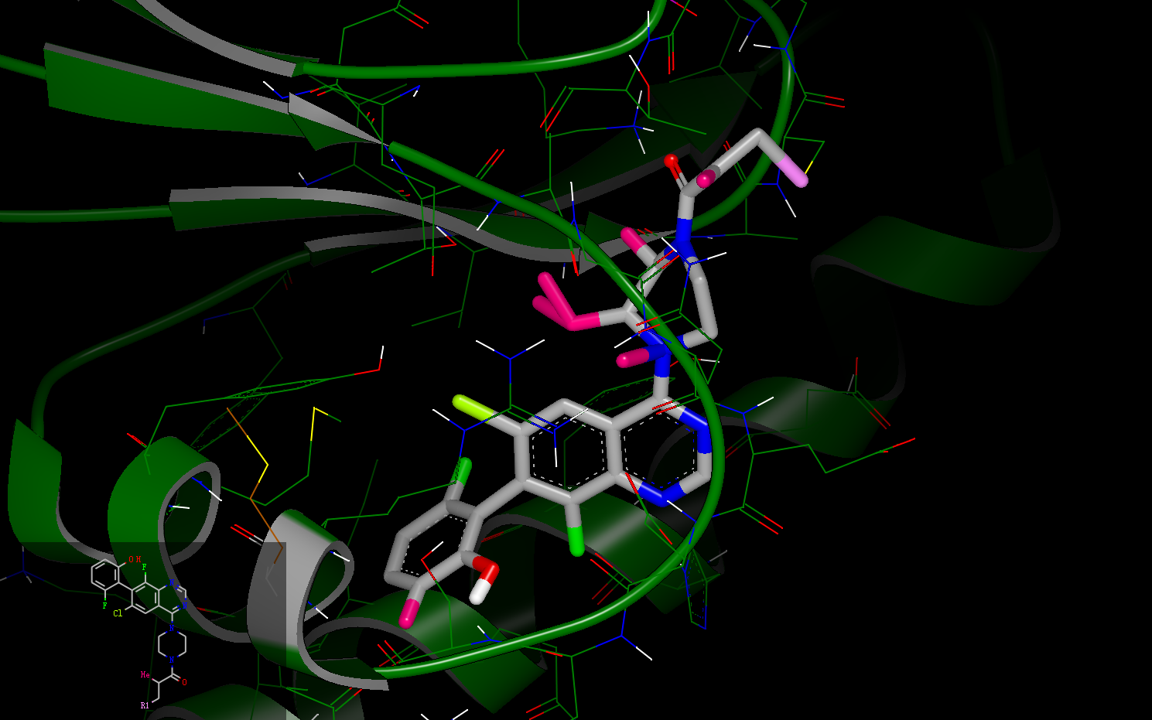
图3S. ARS-1620的极性取代假设
在图2S中,母核与哌嗪环的vdW取代紧挨在一起,这为大环设计提供了足够的提示,如图4S所示。
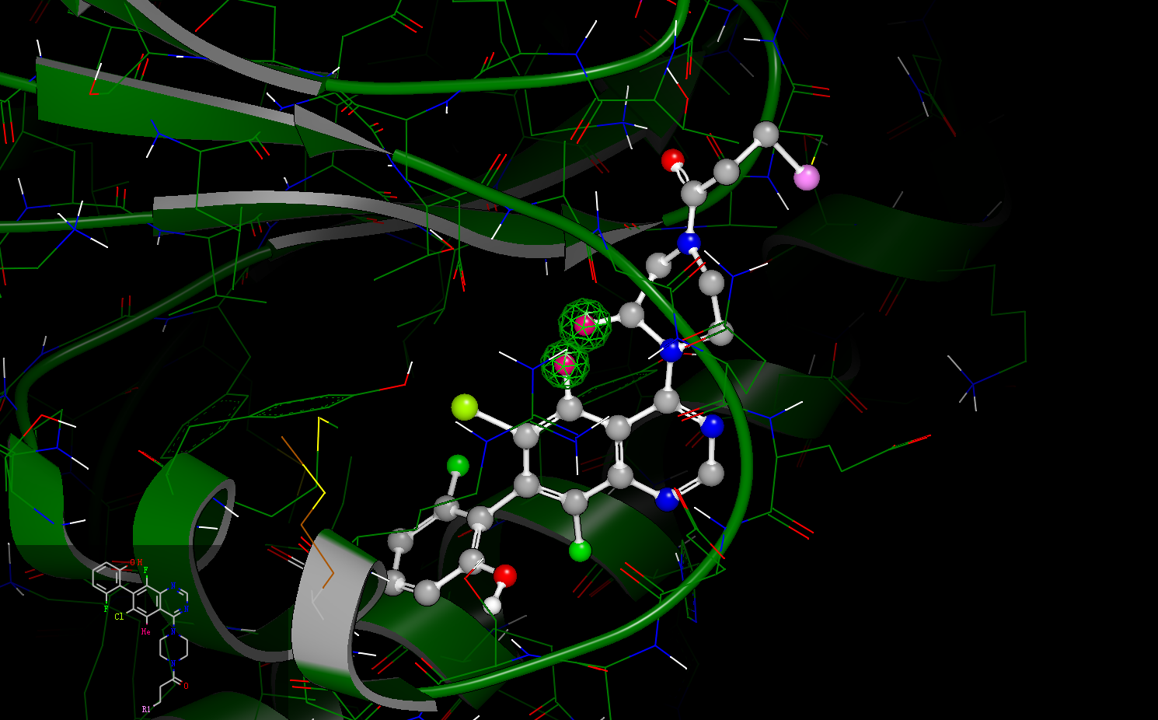
图4S. 母核与哌嗪环的vdW取代假设为大环设计提供了足够的提示
文献
- Wang, X.; Allen, S.; Blake, J. F.; Bowcut, V.; Briere, D. M.; Calinisan, A.; Dahlke, J. R.; Fell, J. B.; Fischer, J. P.; Gunn, R. J.; et al. Identification of MRTX1133, a Noncovalent, Potent, and Selective KRAS G12D Inhibitor. J. Med. Chem. 2022, 65 (4), 3123–3133. https://doi.org/10.1021/acs.jmedchem.1c01688.
- 肖高铿. KRASG12C共价抑制剂MRTX849先导化合物优化过程中的水分子替换. 墨灵格的博客. 2021-08-30. http://blog.molcalx.com.cn/2021/08/30/water-replace-in-mrtx849-lead-optimization.html
- SZMAP. OpenEye. http://www.eyesopen.com/szmap
- Kettle, J. G.; Bagal, S. K.; Bickerton, S.; Bodnarchuk, M. S.; Boyd, S.; Breed, J.; Carbajo, R. J.; Cassar, D. J.; Chakraborty, A.; Cosulich, S.; et al. Discovery of AZD4625, a Covalent Allosteric Inhibitor of the Mutant GTPase KRASG12C. J. Med. Chem. 2022. https://doi.org/10.1021/acs.jmedchem.2c00369.


