
KRASG12C共价抑制剂MRTX849先导化合物优化过程中的水分子替换
摘要:本文以KRAS共价抑制剂MRTX849的先导化合物优化为例,说明水分子替换策略在先导化合物优化过程中的重要性。与经典的分析方法比较发现,分子对接Vina不能区分先导化合物7与引入腈基后12a之间高达440倍的活性差异;也不能区分在萘环8位引入氯与甲基前后在活性上的差异。我们发现非经典的相互作用可以弥补Vina为代表的打分不足,从统计学角度给出合理的SAR解释。GIST与SZMAP计算的热力学分析不仅可以将先导化合物优化最佳位点以优先级最高的方式突显出来,而且还成功地解释先导化合物优化前后的活性差异。
肖高铿/2021-08-30
1. MRTX849的先导化合物优化过程简介
MRTX849 (Adagrasib,图1)是Mariti开发的一种口服有效的、高度选择性的KRASG12C抑制剂。MRTX849通过将KRAS分子不可逆地锁定在其非活性状态来最大化抑制作用,从而防止肿瘤细胞生长并导致肿瘤细胞死亡。与现有的KRAS突变体选择性抑制剂相比,MRTX849的半衰期和进入肿瘤的渗透率显著提高,在整个给药期间中都关停了KRAS信号传导,并显示出更高的抗肿瘤活性。
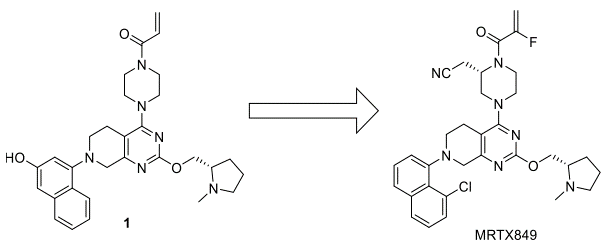
图1. 先导化合物1与临床研究药物MRTX849的结构
MRTX849从先导化合物1优化而来[1]。先导化合物1因为除了发生四氢吡咯环的N-Me发生脱甲基、Michael受体发生GSH加成等代谢反应之外,其萘环羟基容易发生葡萄糖醛酸化与硫酸酯化等II相代谢反应加剧了其代谢不稳定性。CD-1小鼠100mg/kg的PO给药化合物1之后,生物利用度只有2.4%。
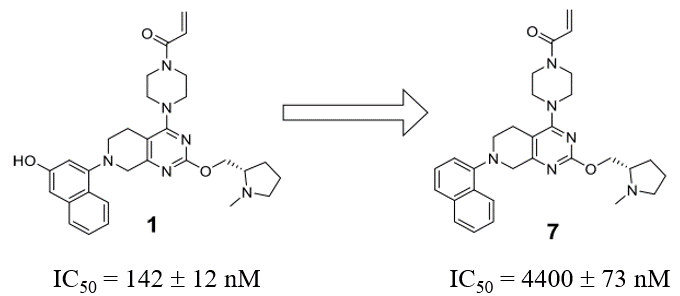
图2. 化合物1及其去羟基衍生物7
为了克服萘环羟基的代谢不稳定性,对其去羟基变得势在必行。根据之前的研究[2],萘环的羟基用来与结合位点的ASP69发生氢键相互作用,1与靶标的复合物结构(PDB 6N2K)也证明了这一点。少了这个氢键相互作用后,衍生物7的细胞活性从化合物1的IC50=142nM降低到IC50=4400nM,这促使研发团队寻求其它位置的修饰以提高活性。
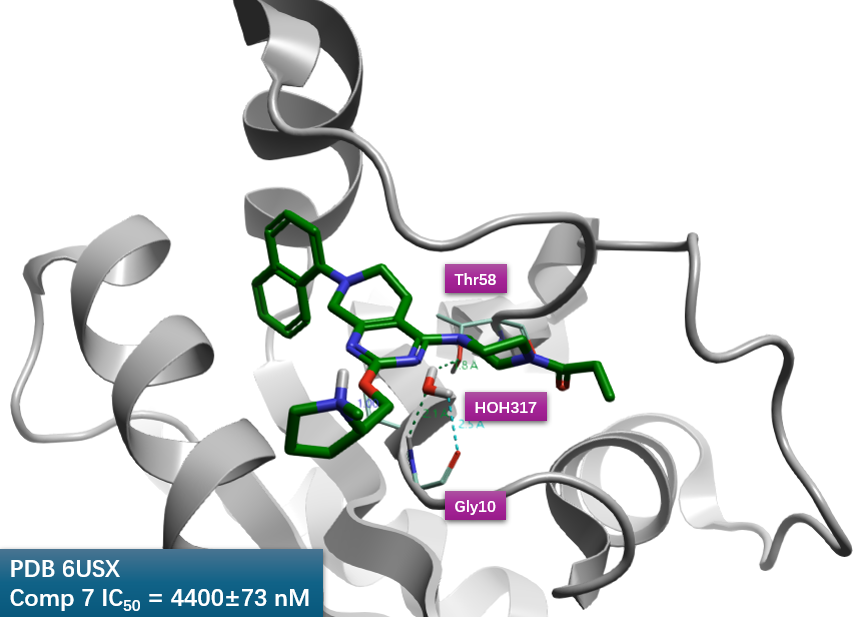
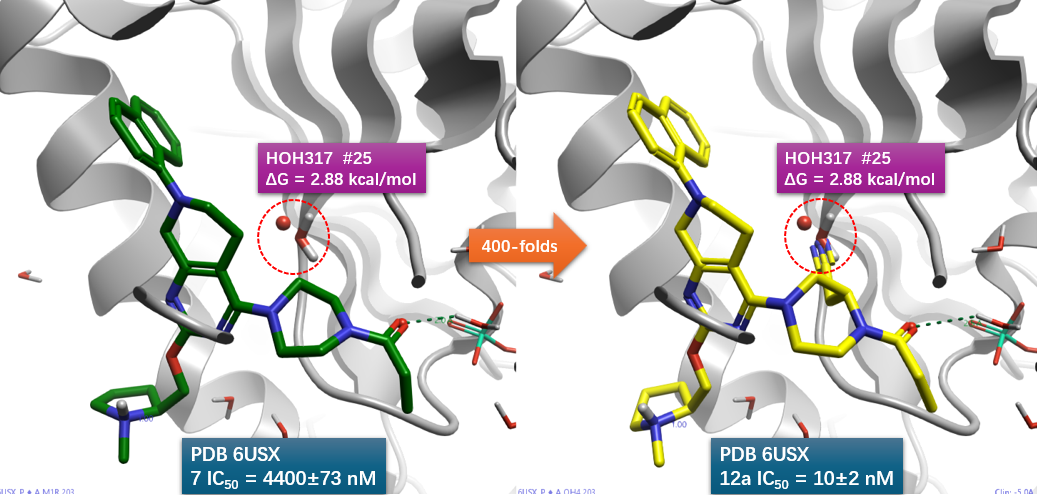
图3. 化合物7-KRAS共晶结构PBD 6USX的HOH317。
根据化合物7与KRAS共晶结构PDB 6USX(图3),在化合物7哌嗪环附近发现了结晶水HOH317,HOH317的氧原子与哌嗪环最近一侧两个碳原子距离分别为3.5与3.7Å;同时结晶水HOH317同时与残基Gly10与Thr58发生氢键相互作用。
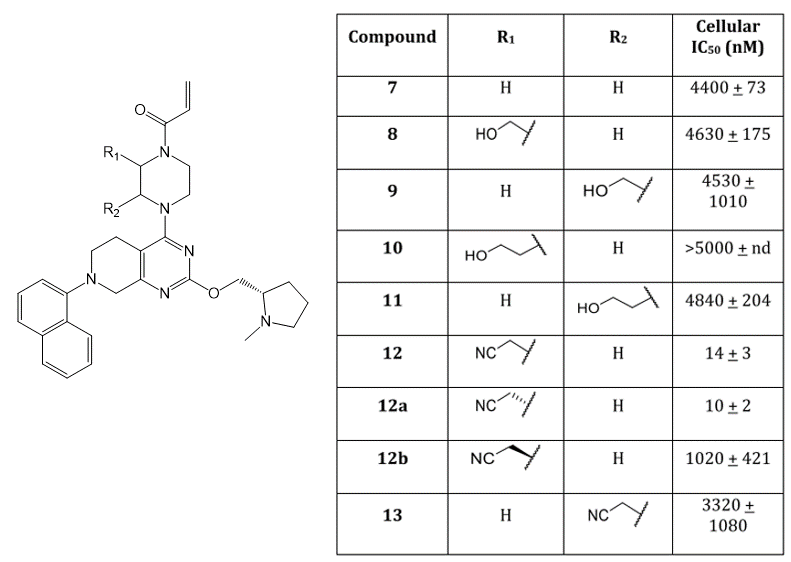
图4. 设计哌嗪取代衍生物以置换结晶水HOH317
已有的研究表明,分析氢键网络,替换合适的水可能会有活性上的增益[3]。于是作者对哌嗪环进行修饰,期望通过结晶水的替换来提高活性。最后合成、测试了图4的系列衍生物。结果表明,R1被乙腈基取代之化后的合物12a,细胞活性从化合物7的IC50=4400nM提高440倍,12a的IC50=10nM。
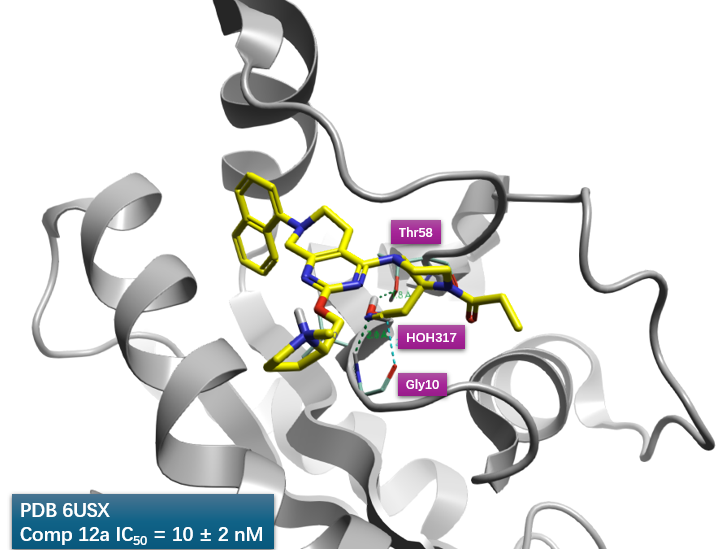
图5. 将12a与KRAS共晶结构PDB 6USZ叠合到7与KRAS的共晶结构PDB 6USX上,在6USX结合位点里比较7,12a的结合模式,发现12a的腈基占据了HOH317的位置。
将12a与KRAS共晶结构PDB 6USZ叠合到7与KRAS的共晶结构PDB 6USX上,比较两者的结合位点,如图5所示,可以发现12a的腈基刚好与HOH317重叠,这说明12a的腈基确实实现了对HOH317的替换。
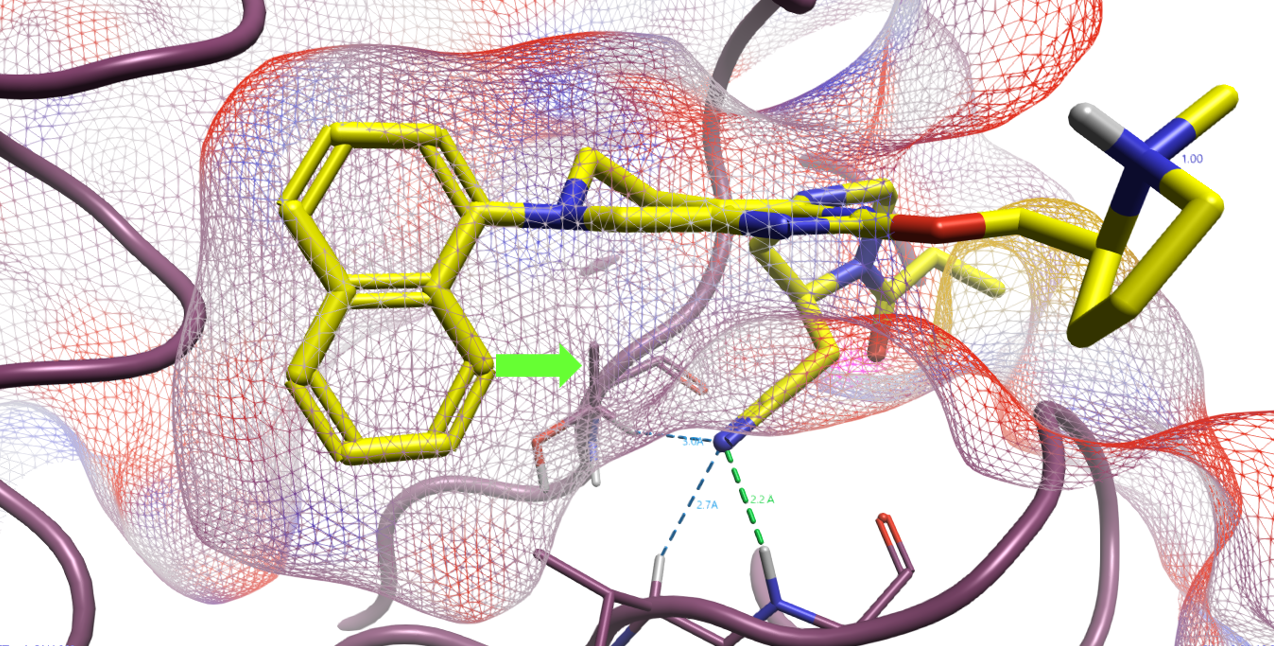
图6. 在12a与KRAS共晶结构PDB 6USZ中,12a萘环片段附近的疏水空隙。
作者还发现PDB 6USZ复合物结构中12a萘环8位的方向上(图6)有一个由Val9、Thr58,、Met72以及Tyr96组成的疏水空隙,因此还对12a的8位用各种疏水的小基团进行了取代,结果如图7所示。甲氧基、腈基取代的化合物活性比12a变差;乙基、三氟甲基取代衍生物活性与12a差不多;而氯(化合物18)与甲基(化合物19)取代的衍生物活性比12a提高约10倍,IC50为1nM左右。
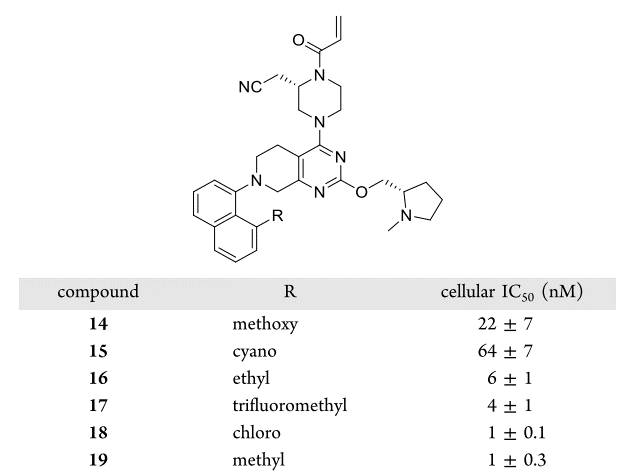
图7. 探索萘环8位对活性的影响
比起化合物1,化合物18、19在CD-1小鼠上表现改善的代谢清除率(IV给药的CL分别为37.3与31.6 mL/min/kg)与口服生物利用度(分别为16.9%与31.1%),因此进一步对18与19进行药效学研究。鉴于18、19良好的活性数据,研究团队启动第2物种的PK研究。在比格犬上,18与19以3mg/kg剂量IV给药后的清除率分别升高到225、151 mL/min/kg,10mg/kg的剂量口服给药后生物利用度仅为4%左右,血浆暴露量很低,仅为AUCinf 小于 0.04 h·μg/mL。根据Xia[4]与Zhao[5]等人的报道,比格犬上差的DMPK数据可能与Michael受体的GSH共轭加成有关,取代的丙烯酰胺比丙烯酰胺对GSH的加成会得以减弱。于是对Michael受体进行修饰,筛选。结果发现在α位上引入F之后的化合物20(即MRTX849)的代谢稳定性提高,小鼠/犬/人全血t1/2分别从18的5/2/14小时提高到大于50/50/50小时。化合物20并非活性最强的化合物(IC50=14nM),却是DMPK等性质综合表现最好的化合物。最终,化合物20作为候选药物进入临床,目前处于3期临床研究阶段。
2. 本文的目
在MRTX849的优化过程中,我们最感兴趣的部分是从7到12a,以及12a到18、19的优化。如何将优化方向以高的优先级引导到正确方向、最终实现优化目标是我们感兴趣的。因此,本文的主要目的是以回溯性的方式探讨几个问题:
- 从化合物7-KRAS共晶结构PDB 6USX出发,如何快速聚焦到HOH317,并提出腈基对HOH317取代方案而设计出化合物12a。
- 从化合物12a-KRAS共晶结构PBD 6USZ出发,如何将对萘环8位的取代提高较高的优先级,并提出氯与甲基的取代方案。
3. 方法
3.1 蛋白的结构准备
将化合物7、12a、20与KRAS共晶结构PDB 6USX、6USZ和6UT0从蛋白质数据库下载到Flare中,并使用来自Protein Prep工具小心地准备,以添加氢原子、优化氢键、消除原子冲突并给蛋白结构分配最佳质子化状态。任何截短的蛋白质链被封端作为蛋白质准备的一部分。使用COBALT多重比对工具在Flare中比对蛋白质序列,随后通过Cα的最小二乘拟合进行叠合。
3.2 GIST分析
了解蛋白活性位点内水分子的行为是药物设计的一个非常重要的方面。在蛋白质-配体复合物中,了解桥连水分子的稳定性是决定药物设计策略的关键。如果水分子不是特别稳定,可以设计配体以取代它,并与蛋白质活性位点直接相互作用:这通常会导致配体生物活性的增加。
GIST是一种水热力学性质分析方法,用于评估结合口袋的水合作用,并通过在分子动力学运行结束时对显式溶剂分布采样来计算相关的水热力学性质。GIST分析的结果显示为活性位点水合作用的三维等值面映射“happy”(绿色,与负∆G相关)和“unhappy”(红色,与正∆G相关)区域。unhappy区域映射的是蛋白质活性位点内更可能的可成药区域。happy区域映射的是蛋白结合位点内较低可能成药的区域,在那里水分子更稳定,因此更难置换。
在本文中,用Flare分对PDB 6USZ Apo结构(不包含配体、金属离子与结晶水)进行了GIST分析,使用了如下条件:
- Calculation method: Normal
- Ligand: None
- Grid spacing:0.5 Å
- Grid Definition:Ligand
- Chains: 不包含水、配体与其它
- Simulation length: 20ns
- Solvent Model: explicit TIP4Pew Water
4. 结果
4.1 比较7、12a、18、19、20(MRTX849)与KRAS的相互作用
分子对接是大家喜欢用来对感兴趣化合物进行分析与排序的工具,因此用经典的分子对接软件Vina(Version 1.2)对化合物进行打分,以考察分子对接能否区分化合物的活性差异。在本算例中,将化合物从复合物结构中提取出来,并将配体的Michael受体部分还原为丙烯酰胺。化合物18、19的结合模式由6UT0的配体修改而来。将蛋白部分按两种方式处理:1)保留Cys12为Cys的形式,但是会与配体的丙烯酰胺发生碰撞;2)将Cys12突变为Ala12(C12A),以消除Cys12巯基与配体丙烯酰胺的碰撞。然后将配体与对应的蛋白结构用经典的AutoDock Vina 1.2进行打分,结果如表1所示。
| Comp ID | PDB ID | Vina Scorea(kcal/mol) | Vina Scoreb(kcal/mol) | Hydrophobicc | H-Bondc |
| 7 | 6USX | -6.99 | -10.84 | 63.97 | 2.37 |
| 12a | 6USZ | -6.97 | -9.59 | 60.94 | 2.59 |
| 18 | 6UT0 | -6.70 | -10.41 | 60.54 | 2.37 |
| 19 | 6UT0 | -7.03 | -10.46 | 63.97 | 2.36 |
| 20 | 6UT0 | -7.09 | -10.55 | 58.43 | 2.37 |
a. KRAS-Cys12蛋白结构;b. Cys12突变为Ala12的蛋白结构;c.Hydrophobic与H-Bond分别为(Cys12突变为Ala12的蛋白结构时)Vina加权前的相应打分项贡献值。
结果表明,Vina 1.2分子对接打分不能解释先导化合物7与在哌嗪环上引入腈基后12a之间IC50上440倍的差异;也不能解释12a与在其萘环上引入疏水基团氯与甲基而活性得到提升的18、19之间的IC50差异。在本算例中经典的分子对接Vina打分不能为先导化合物优化提供直接的指导。
比较7与12a的加权前疏水与氢键两个贡献值可发现,由于乙腈基加入,疏水项贡献略有降低、氢键项略有升高,但是对预测的ΔG没有往正确的方向走。与12a相比,18与19分别在12a的萘环上引入了疏水的氯与甲基,可以发现Vina打分对引入氯并不敏感,疏水项贡献分别为60.94与60.54,几乎没有变化;而对引入甲基更加敏感,疏水项贡献分别为60.94与63.97。腈基与氯的贡献被AutoDock Vina的打分严重低估。
被Vina打分严重低估的腈基与KRAS之间的相互作用,可用Kuhn等人[6]所述的非经典相互作用来解释。如图8所示,腈基氮与Gly10的酰胺氮距离为3.2Å(与酰胺氮上氢的距离为2.2Å),两者之间满足CN…N_pi_don相互作用模式,此模式RF=1.85,是一种非常高频的相互作用。腈基氮还与在Val9、Thr58中与极性原子连接的C-H发生CN…C_ali_don模式的相互作用,此模式RF=1.71,也是非常高频的一种相互作用。腈基与KRAS之间发生的CN…N_pi_don与CN…C_ali_don高频相互作用模式,可以用来解释腈基引入而提高活性400多倍的现象。
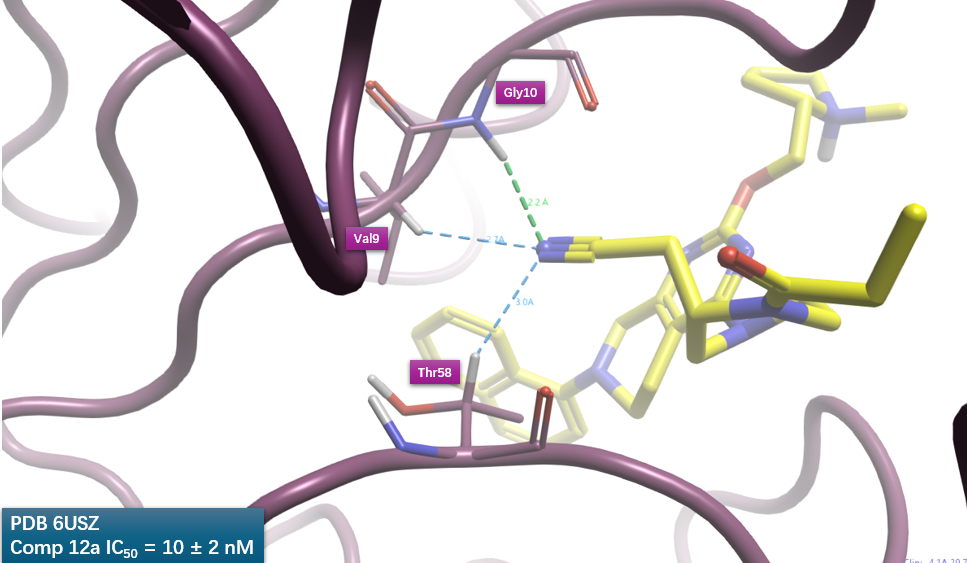
图8. 化合物12a的腈基与KRAS之间非经典相互作用分析
被Vina打分低估的氯与KRAS之间的相互作用,也可用Kuhn等人[6]所述的非经典相互作用来解释。如图9所示,氯与Val9以及Met72的末端甲基发生接触,满足Cl…C_ali_apol相互作用模式,此模式RF=1.13,是一种高频的相互作用。氯还与Try96的两个苯环碳发生Cl…C_pi_phenyl模式的相互作用,此模式RF=1.37,也是一种高频的相互作用。氯与KRAS之间发生的Cl…C_ali_apol与Cl…C_pi_phenyl高频相互作用模式,可以用来解释12a引入氯而提高活性的现象。
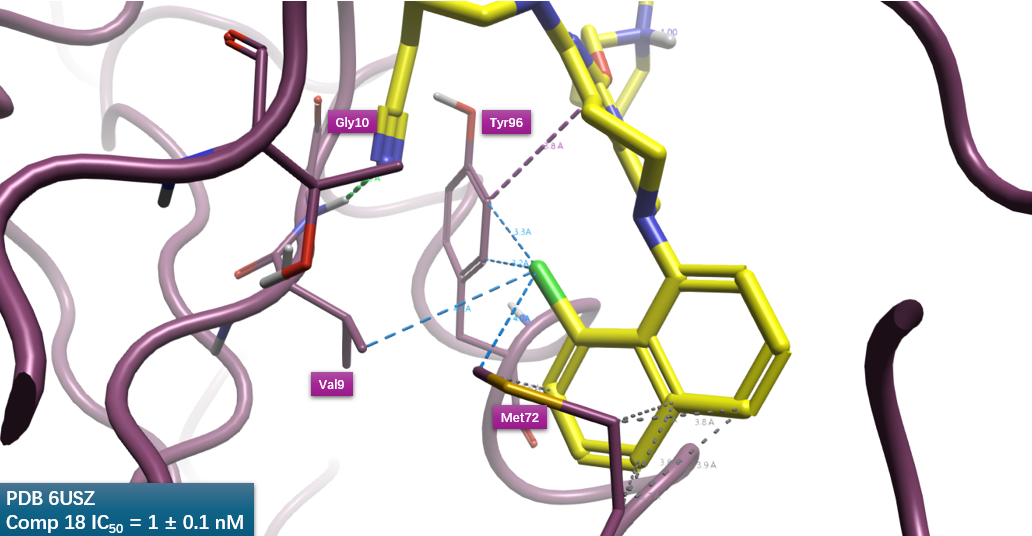
图9. 化合物18萘环的8位氯与KRAS之间的非经典相互作用
总的来说,Vina打分为代表的经典相互作用分析不能解释MRTX649先导化合物优化过程中引入腈基与甲基改进活性中的用途,而腈基与氯满足高频非经典相互作用模式,可以从统计学角度用非经典相互作用来解释SAR。巧合的是,这些非经典相互作用均发生在结合口袋apo-结构的高能水合位点,详见后续的apo-GIST分析。
4.2 GIST分析化合物7-KRAS共晶结构PDB 6USX的结合位点
“可成药的”结合位点通常被水分子占据,由于焓和熵的原因,水分子在能量上更倾向于在大量水中。在蛋白质-配体复合物中,了解桥连水分子的稳定性是决定药物设计策略的关键。如果水分子不是特别稳定,可以设计配体以取代它,并与蛋白质活性位点直接相互作用:这通常会导致配体生物活性的增加。
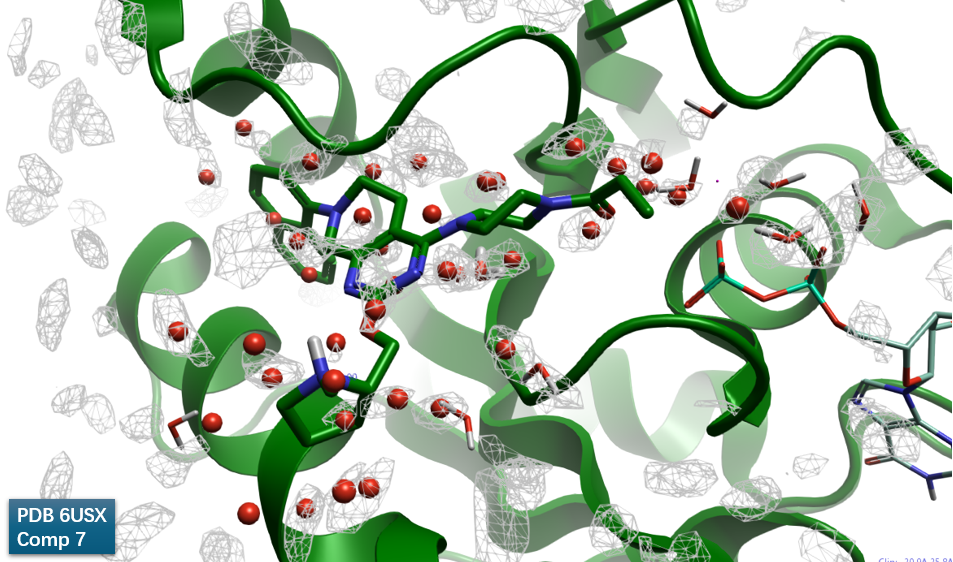
图10. 6USX结合位点处GIST计算结果。其中灰色网格:Water Density\(>= 4\); 绿色飘带: KRAS;红色球状:GIST预测的水合位点(仅显示配体 4 Å 之内点)。
对7-KRAS共晶结构Apo结合位点的GIST分析,结果如图10所示,结合位点基本被unhappy的水合位点覆盖,这是一个从水能量学角度讲成药性好的结合位点。我们对靠近配体的两个高能水合位点感兴趣,如图11所示:其一是水合位点#25,靠近哌嗪环,与共晶水分子HOH317位置重合,\(\Delta G = 2.88 kcal/mol\);另一个水合位点#59,靠近萘环,其\(\Delta G = 3.67 kcal/mol\)。对这个两个高能水合位点的置换设计,有望提高化合物的结合亲和力。这两个水合位点距离化合物7大约1-2键范围内的等值图,在这个距离范围内容易设计简单基团对水分子进行置换而达到先导化合物亲和力优化的目的。
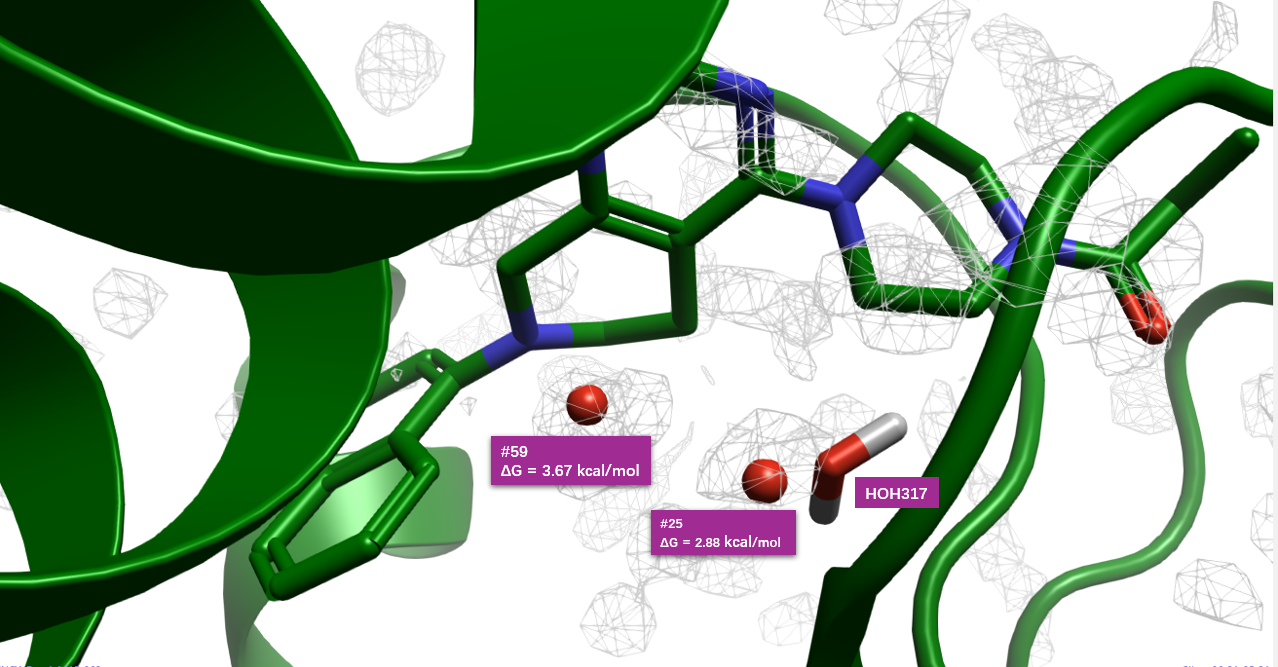
图11. 在化合物7-KRAS共晶结构PDB 6USX的结合位点处,两个靠近配体的关键水合位点示意图
将化合物 20-KRAS 共晶结构(PDB 6UT0)与化合物 7-KRAS 共晶结构(PDB 6USX)进行结构叠合,分析并比较化合物 20(MRTX849)与 PDB 6USX 中结合口袋内关键水合位点之间的空间分布与相互作用关系,如图12所示:化合物20的萘环氯原子与哌嗪侧链上的腈基分别置换了高能水合位点#59与#25。这可部分解释化合物20相对于7的400倍的活性提升。这证明了GIST水合自由能计算对指导先导化合物优化的巨大价值。
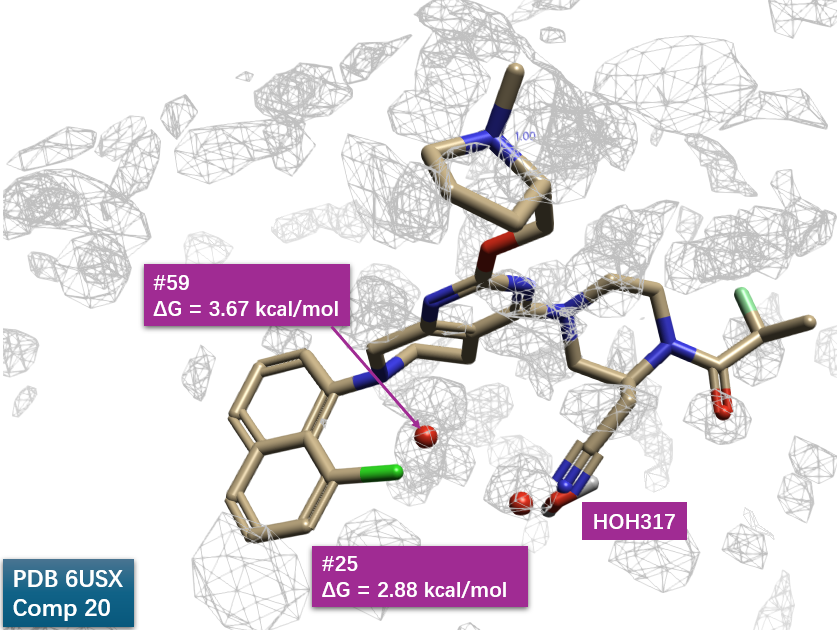
图12. 通过将化合物 20 与 KRAS 的共晶结构(PDB 6UT0)叠合至化合物 7-KRAS 共晶结构(PDB 6USX),分析了化合物 20(MRTX849)与位于 PDB 6USX 结合口袋中关键水合位点之间的关联
总的来说,GIST计算的高能水合位点可以将先导化合物的优化位点以最高的优先级标注出来,并提示采用水分子替换/置换策略可提高化合物的活性,为药化工作提供可靠的指导。
4.3 用SZMAP稳定化自由能与中性探针自由能差来探讨KRAS G12C共价抑制剂的SAR
关于稳定化自由能与中性探针自由能差的计算概念、原理与应用,请参见我的另一篇博客介绍:水分子的稳定化自由能计算及其应用。该文已经详细的介绍了在MRTX849系列化合物SAR研究的应用,这里只是给出个结论。
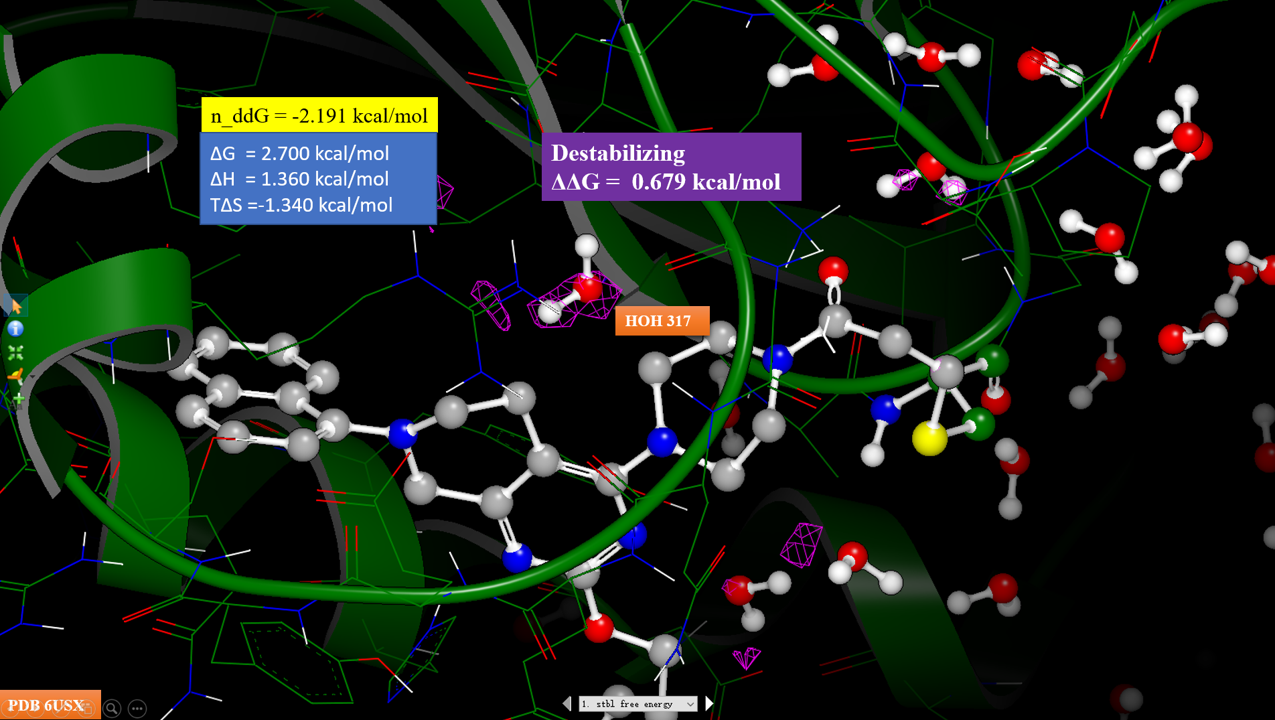
图13. 化合物7与KRAS共晶结构(PDB 6USZ)的稳定化自由能计算结果。紫色等值图:ΔΔG大于0.5kcal/mol区;黄色等值图:ΔΔG小于-0.5kcal/mol区。
对化合物7与KRAS共晶结构PDB 6USZ进行稳定化自由能计算并将结果展示为图13所示等值图。可以发现,HOH317处于紫色的正稳定化自由能区,这意味着HOH317对化合物7与KRAS的结合不利,应该予以替换或修饰配体以改善与水的相互作用。
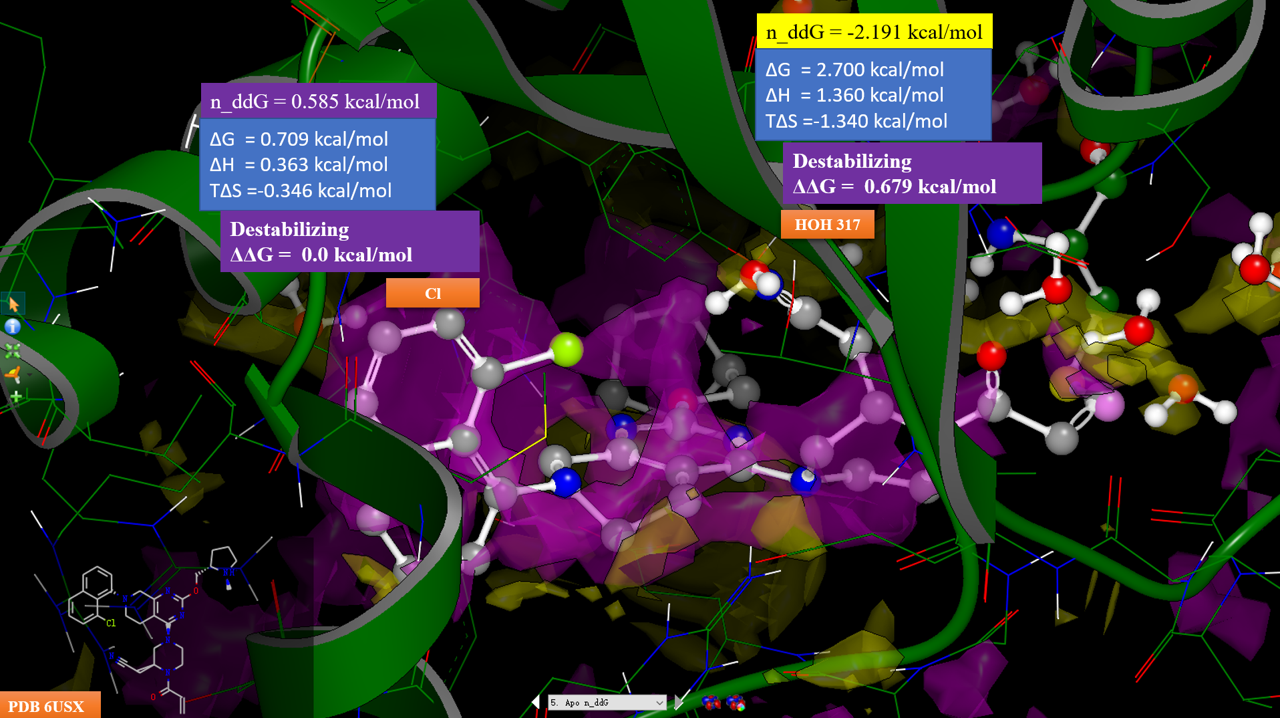
图14. 化合物7与KRAS共晶结构(PDB 6USZ)Apo结构的中性探针自由能差计算结果。紫色等值图:n_ddG大于0.5kcal/mol区;黄色等值图:n_ddG小于-0.5kcal/mol区。配体为化合物18,其构象是将PDB 6UT0按CΑ叠合到PDB 6USX之后修改其中的配体MRTX849而来。
对6USX的Apo结构(不包含配体、水、金属)进行中性探针自由能差分析,等值图分布如图14所示,紫色为配体极性基团不利于活性区域,黄色为配体极性基团有利于活性区域,反之相反。可以发现,HOH317水被一个黄色的区块覆盖,且n_ddG = -2.191 kcal/mol,这说明该位点应该用一个极性基团对HOH317进行替换。为了验证这一点,将MRTX849与KRAS的复合物结构PDB 6UT0叠合到6USX上,并将6UT0配体的氟原子修改为氢而得到化合物18的构象。叠合之后,我们可以发现,哌嗪环上的乙腈基氮原子与水叠合在一起。因此我们可以确认,化合物12a、18的哌嗪环腈基对HOH317的替换是活性提升440倍的关键。
从图14还可以看到,化合物18萘环氯原子刚好落在紫色的区块里,氯原子位置对应坐标的n_ddG = 0.585 kcal/mol,这说明氯是靠疏水效应踢走“可能”的水而在活性上获益,化合物18因此比12a的活性提高10倍。实际上,这也与图7的事实一致:在12a萘环8位引入三氟甲基、甲基、乙基等疏水基团都是对活性有利的,而引入腈基与甲氧基则对活性不利。这进一步证明了n_ddG给出的SAR信息具有非常高的价值。
4.4 Michael受体共价结合片段羰基氧与Happy结合水的相互作用
观察到在结合位点的Zn2+离子与4个水、辅因子形成了金属配位相互作用,如图15所示。4个与Zn离子配体的水(HOH304、307、309、319)具有相对高的密度(\(density >= 4\)),而且均为Happy water,它们的GIST水合自由能(\(\Delta G\))分别为-0.77、-1.67、-1.50、-1.35 kcal/mol。其中HOH319与#2水合位点重合,介导了Michael受体羰基氧与Zn离子之间的氢键相互作用,这进一步有利于配体与蛋白的相互作用。
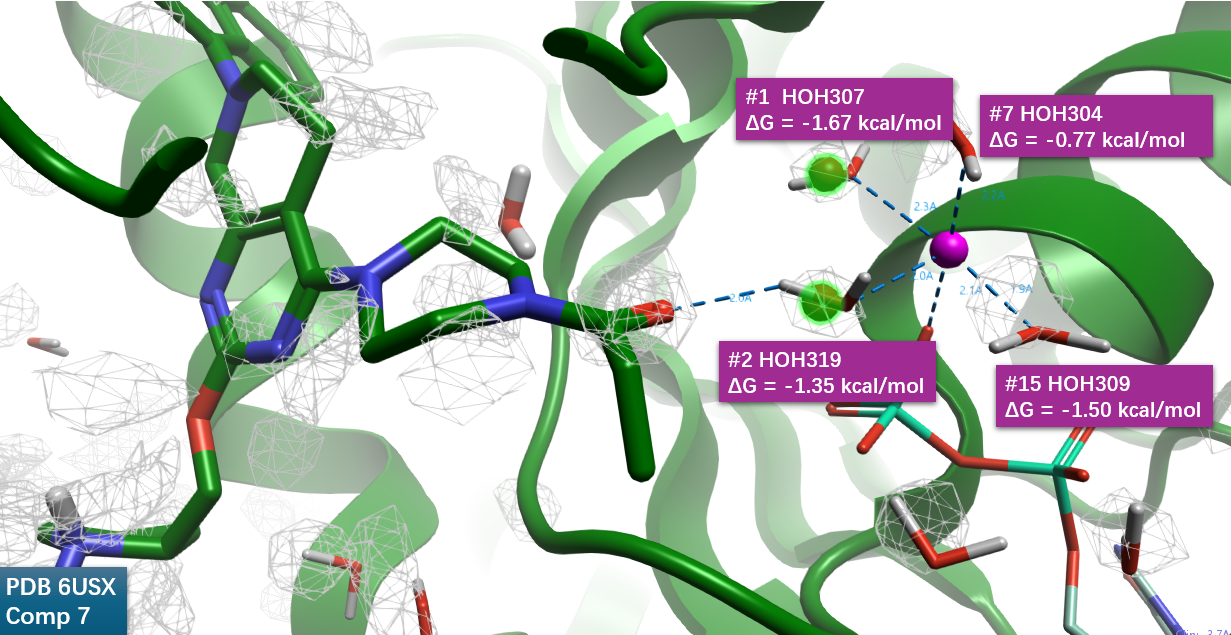
图15. Michael受体共价结合片段羰基氧与Happy结合水的相互作用
5. 小结
本文以KRAS共价抑制剂MRTX849的先导化合物优化为例,说明水分子替换策略在先导化合物优化过程中的重要性。
经典的分析方法分子对接Vina打分不能区分先导化合物7与引入腈基后12a之间高达440倍的活性差异;也不能区分在萘环8位引入氯与甲基前后在活性上的差异。我们发现非经典的相互作用可以弥补Vina为代表的打分不足,从统计学角度给出合理的SAR解释。
GIST计算的热力学分析不仅可以将先导化合物优化最佳位点HOH317以及萘环8位以优先级最高的方式突显出来,而且还成功地解释先导化合物优化前后的活性差异。SZMAP的稳定化自由能进一步确认HOH317对活性不利,中性水水探针自由能差结果建议用极性基团对HOH317进行替换、并建议对萘环8位用疏水基团替换,这与化合物的活性趋势事实一致。
GIST分析与SZMAP的中性探针自由能差分析,是两种互补的方法:GIST的高能水分子位置直接提示了修饰位置;中性探针自由能差则给出了具体的修饰方向。两者应该结合使用,加速药化项目的进度。
6. 文献
- Fell, J. B.; Fischer, J. P.; Baer, B. R.; Blake, J. F.; Bouhana, K.; Briere, D. M.; Brown, K. D.; Burgess, L. E.; Burns, A. C.; Burkard, M. R.; et al. Identification of the Clinical Development Candidate MRTX849 , a Covalent KRAS G12C Inhibitor for the Treatment of Cancer. J. Med. Chem. 2020, 63 (13), 6679–6693. https://doi.org/10.1021/acs.jmedchem.9b02052.
- Fell, J. B.; Fischer, J. P.; Baer, B. R.; Ballard, J.; Blake, J. F.; Bouhana, K.; Brandhuber, B. J.; Briere, D. M.; Burgess, L. E.; Burkard, M. R.; et al. Discovery of Tetrahydropyridopyrimidines as Irreversible Covalent Inhibitors of KRAS-G12C with in Vivo Activity. ACS Med. Chem. Lett. 2018, 9 (12), 1230–1234. https://doi.org/10.1021/acsmedchemlett.8b00382.
- Michel, J.; Tirado-Rives, J.; Jorgensen, W. L. Energetics of Displacing Water Molecules from Protein Binding Sites: Consequences for Ligand Optimization. J. Am. Chem. Soc. 2009, 131 (42), 15403–15411. https://doi.org/10.1021/ja906058w.
- Xia, G.; Chen, W.; Zhang, J.; Shao, J.; Zhang, Y.; Huang, W.; Zhang, L.; Qi, W.; Sun, X.; Li, B.; Xiang, Z.; Ma, C.; Xu, J.; Deng, H.; Li, Y.; Li, P.; Miao, H.; Han, J.; Liu, Y.; Shen, J.; Yu, Y. A chemical tuned strategy to develop novel irreversible EGFR-TK inhibitors with improved safety and pharmacokinetic profiles. J. Med. Chem. 2014, 57, 9889−9900.
- Zhao, B.; Xiao, Z.; Qi, J.; Luo, R.; Lan, Z.; Zhang, Y.; Hu, X.; Tang, Q.; Zheng, P.; Xu, S.; Zhu, W. Design, synthesis and biological evaluation of AZD9291 derivatives as selective and potent EGFRL858R/T790M inhibitors. Eur. J. Med. Chem. 2019, 163, 367−380.
- Kuhn, B.; Gilberg, E.; Taylor, R.; Cole, J.; Korb, O. How Significant Are Unusual Protein–Ligand Interactions? Insights from Database Mining. J. Med. Chem. 2019, 62 (22), 10441–10455. https://doi.org/10.1021/acs.jmedchem.9b01545.


