
SHP2别构抑制剂TNO155发现过程回顾与基于结构的SAR分析
摘要:NO155是诺华开发的First-In-Class选择性、可口服的SHP2别构抑制剂,具有理想的成药性值,是首个进入临床研究的别构小分子SHP2抑制剂。本文回顾了TNO155的先导物发现与优化过程,并对关键的苗头到先导以及先导化合物优化进行了基于结构的SAR分析。
肖高铿/2021-09-07
1. TNO155简介及本文的目的
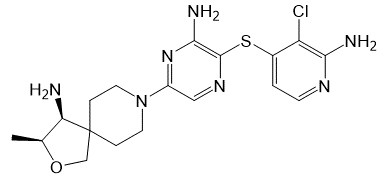
图1. TNO155的化学结构式
TNO155化学结构式见图1,是诺华开发的first-in-class选择性、口服SHP2别构抑制剂,具有理想的成药性值(比如高透过性、高溶解度、不抑制CYP450、理想的临床前PK参数),TNO155是首个进入临床研究的别构小分子SHP2抑制剂。
本文的主要目的是:
- 回顾TNO155的发现过程,了解SHP2抑制相关的SAR。
- 尝试复盘苗头到先导、以及先导优化过程的SAR。
尝试用Flare的可视化技术、XED场技术、水热力学性质计算等基于结构的设计方法(SBDD)来分析TNO155发现过程中苗头到先导(Hit-2-Lead)与先导化合物优化(Lead Optimization)的SAR,并利用SBDD给出的SAR信息来加速药物发现进程。
2. TNO155发现过程回顾

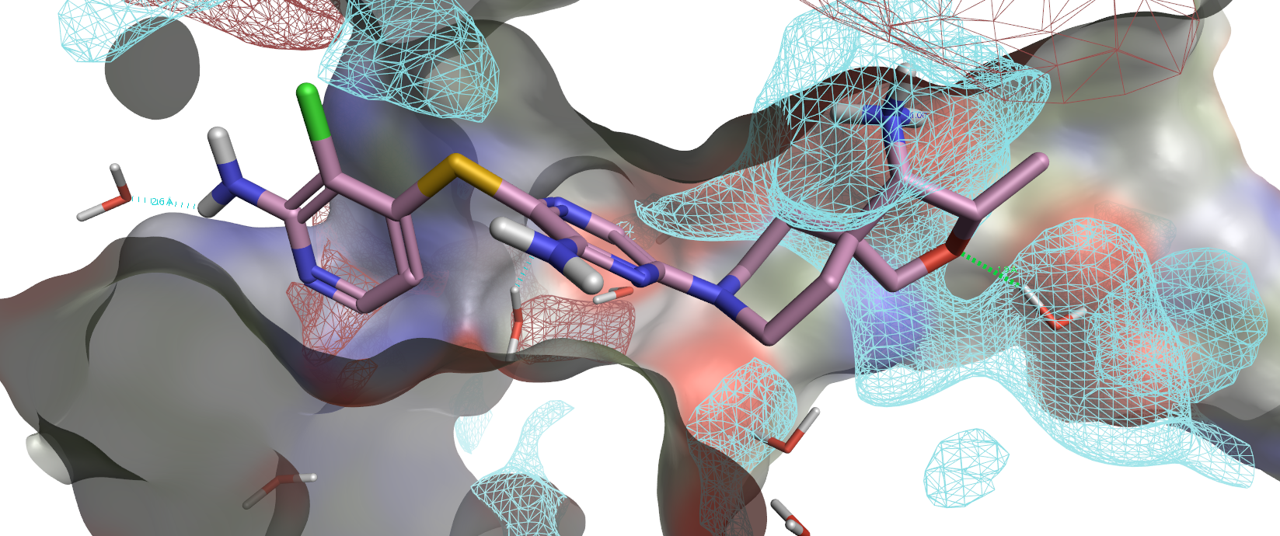
图2. A. 氨基嘧啶类苗头化合物2。 B.2与SHP2共晶结构PDB 5EHP是一个关闭、非活性的构象,分辨率为1.8Å。绿色:N-端SH2结构域;蓝色:C-端SH2结构域;黄色:PTP结构域。放大的小口腔:三个结构域交汇处的别构结构位点。C. 与化合物2结合的别构位点残基注释。图片来源:文献[1]。
在一个对150万化合物的高通量筛选中,诺华发现了苗头化合物2(图2. SHP386),一个新颖的小分子别构抑制剂,可以让SHP2稳定地保持非活性构象。
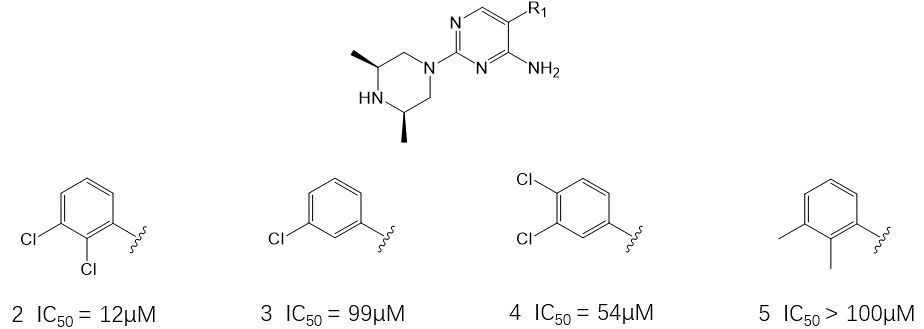
图3. 芳环部分的初步SAR
对氨基嘧啶类苗头化合物2的优化是从保持哌嗪环不变而探索芳环SAR开始:删除邻位氯(化合物3)、将邻位氯迁移到对位(化合物4)以及将两个氯用生物等排体甲基替换(化合物5)均降低了生化抑制活性,如图3所示。鉴于此,在本优化阶段中认为二氯苯基是必须的,因为其高效地填充了由ARG111, THR253, LEU254, GLN257以及GLN495组成的疏水口袋,如图2 C所示。
如图2C所示,苗头化合物2的哌嗪环片段占据了别构结合位点内由PHE113、HIS114、GLU249、GLU250以及THR218等残基组成的极性腔,同时也暴露于溶剂中。由此,作者假设将氨基往这些极性残基延伸使得发生新的相互作用成为可能,并增加对磷酸酯酶的抑制活性。
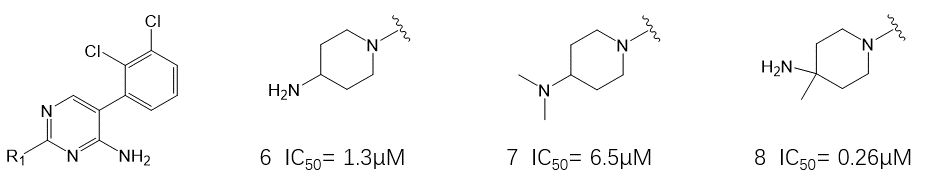
图4. 氨基片段的初步SAR
接下来保留二氯苯基不变,而主要对哌嗪环进行修饰,如图4所示。哌嗪环用4-氨基哌啶替换后的化合物6活性提高了10倍(IC50=1.3µM);对氨基的烷基化则对活性不利,比如化合物7的IC50=6.5µM;如果在哌啶环上氨基偕位增加一个甲基,使氨基处于稳定的平伏键上,比如化合物8,则提高活性至IC50=0.26µM。更重要的是,化合物8在细胞水平(KYSE-520模型)显示出对p-ERK中等程度的调控活性,IC50=1.98µM。
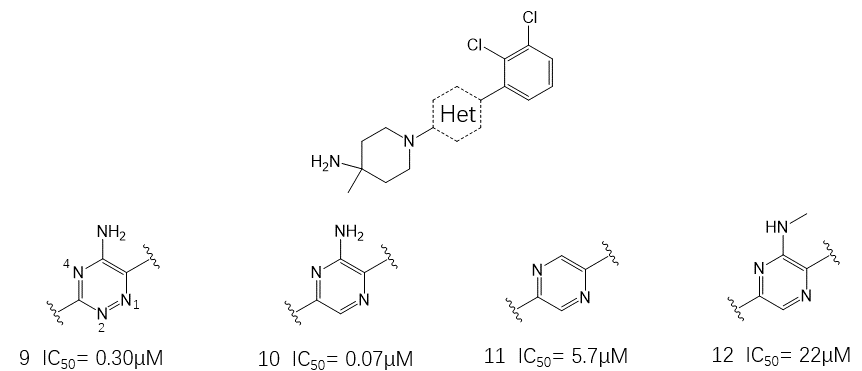
图5. 杂环母核的初步SAR
随着对芳基与氨基片段优化的初步完成,作者将重点放到了中心的嘧啶环上,如图5所示。首先保留氨基与E250的相互作用,发现1,2,4-三嗪保留了抑制活性,化合物9的IC50=0.30µM。作者认为三嗪环的N-1总是朝向ARG111,删除N-2可保留活性。并假设,增加氮的碱性可增强与ARG111之间的相互作用。基于这个假设,测量了质子化三嗪与吡嗪环的pKa分别为4.7与2.9。结果发现,吡嗪环显著地的提高了酶学活性(10的IC50=0.07µM)与KYSE-520细胞活性(p-ERK IC50=0.250µM)。去掉氨基的化合物11与在氨基上增加立体位阻的化合物12均降低了活性,这可能与它们干扰了与GLU250相互作用有关系(参见下文)。
SHP099(10)被证明是一种中等强度的SHP2抑制剂,其酶学IC50=0.07µM,食道癌模型KYSE-520细胞水平p-ERK IC50=0.25µM,抗增殖活性IC50=2.5µM。高水溶性、选择性和口服生物利用度使 10 的体内表征成为可能,并在鼠异种移植瘤模型中证明是有效的。10 还提升了肿瘤中CD8+ IFN-γ+ T 细胞,减少了 CT-26 荷瘤小鼠的肿瘤负荷,并在异种移植瘤模型中与 PD-1 阻断协同作用。 然而,10 在体外3T3 NRU光毒性试验中证明具有光毒性(PIF:219;照射IC50 = 4.5µM),口服给药后的小鼠局部淋巴结光照试验(口服photo- LLNA)表明在药理学相关暴露水平下光毒性具有剂量依赖性。这可能是由于强 UV/Vis 吸光 (Emax352 nm = 14300 M-1cm-1)的发色团所致。此外,大鼠每日给药治疗 2 周后,由于磷脂质沉积症,导致肝细胞和肝内Kupffer细胞出现空泡化,电子显微镜证实了这一点。10 还与 hERG 通道结合并抑制 (IC50 = 5.9 µM),这增加了与化学类型相关的心血管风险。我们将这些安全性发现归因于较大的 Vdss(7 L/Kg,大鼠)和 胺基 pKa(9.5 ),这与 10 的两亲性、阳离子性质一致。总而言之,10 中观察到的光毒性、磷脂沉着症和选择性问题为进一步的优化提供了目标。
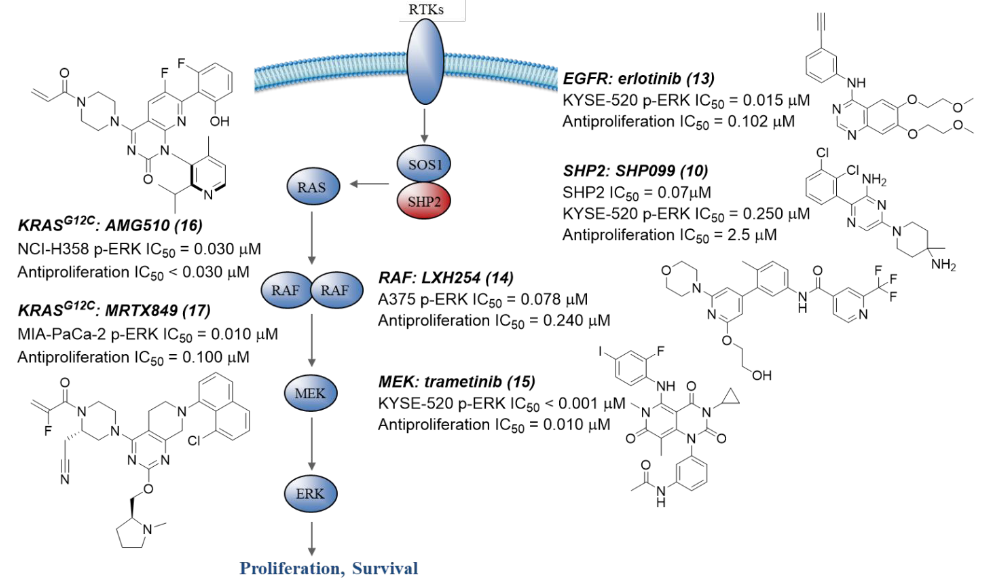
图6. RTK-RAS-MAPK通路上的小分子抑制剂。图片来源:文献[1]。
除了最初的安全问题外,与其他临床使用的RTK和MAPK通路抑制剂相比,10的通路抑制活性也被证明较差。例如,在KYSE-520细胞模型上,已上市的EGFR抑制剂厄洛替尼(13,图6)调节p-ERK的IC50 = 0.015 µM,抑制增殖的IC50 = 0.102 µM,相比之下10的活性低了约10倍,p-ERK IC50 = 0.250 µM,抗增殖 IC50 = 2.5 µM。此外,还通过SHP2下游节点对MAPK通路进行了令人印象深刻的调节。RAF抑制剂 LXH254 (14)、曲美替尼 (15,一种MEK抑制剂) 和临床使用的ERK抑制剂均在不到0.250 µM浓度下对细胞增殖(A375 和 KYSE-520)的抑制率达到50%,总体上优于10的抗增殖IC50 = 2.5 µM。最近报道的靶向突变KRAS的抑制剂也具有类似的药效趋势(例如,AMG510(16):p-ERK IC50 = 0.030 µM,抗增殖IC50小于0.030 µM,NCI-H358模型;MRTX849(17):p-ERK IC50 小于 0.010 µM,抗增殖IC50大于0.100 µM,MIA-PaCa-2 模型)。与上述临床使用的MAPK调节剂类似,我们设想通过 SHP2 抑制实现近乎完全的通路抑制对于作为单药用药的最大临床效果是必要的。在最小化暴露的同时最大化药效可能会提供更有用的治疗指数。因此,有必要进一步优化10以抑制SHP2,同时保持10的理想理化性质和选择性,并避免化学骨架的不良毒性,如光毒性、磷脂沉着症和hERG抑制。
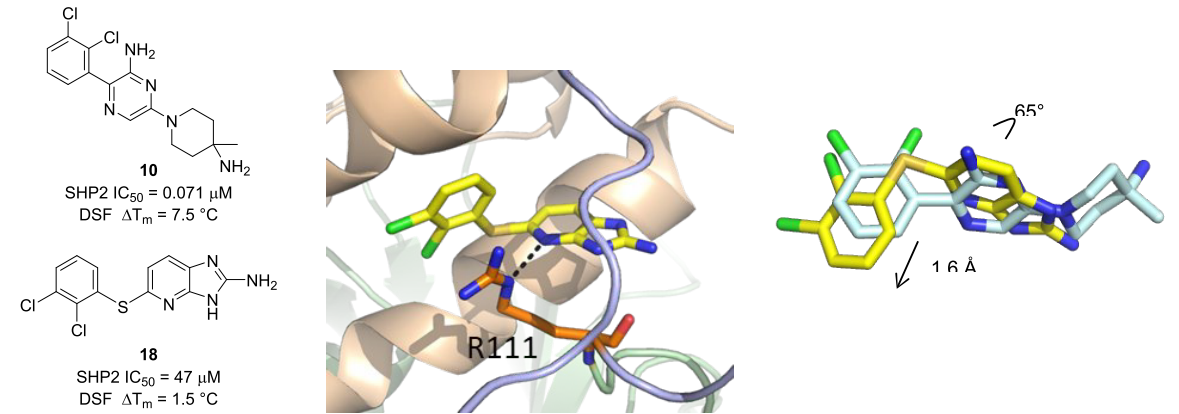
图7. A. 氨基吡嗪10与氮杂苯并咪唑类高通量筛选苗头化合物18。B. 18与SHP2共晶结构PDB 6MDD. C. 18的结合构象(蓝色)与10的结构构象(灰色)叠合比较,中心的杂芳环与一端的芳香环发生了偏移。图片来源:文献[1]。
在对吡嗪环系列进行SAR开发的时候,另有一个高通量筛选结果影响了既定的活性优化策略。比如氮杂苯并咪唑类化合物18(图7 A)可以微弱的抑制SHP2,酶学IC50 = 47 µM。如图7B所示,18与10具有相似的SHP2结合模式,共晶结构表明18的氮杂苯并咪唑与10的吡嗪占据同一个蛋白通道的相似区域。18的吡啶环与ARG111发生氢键相互作用,二氯苯基的芳环与ARG111发生阳离子-π堆积相互作用。这让人联想到ARG111与10吡嗪N之间的相互作用。与10明显不同的是18的硫醚。如图 4C所示,与10相比,18的芳基-S-芳基桥迫使中心环平面偏移了65°。此外,18 中S原子使二氯苯环移动了约 1.6 Å。出于对这种相关却又受干扰的结合模式的兴趣,作者将硫醚结构迁移到吡嗪系列并考察其对活性的影响。

图8. 硫醚连接臂的SAR
最初的二氯硫醚类似物24(图8)体外活性比10更强,酶学IC50 = 0.029 µM,KYSE-520细胞模型p-ERK IC50 = 0.195 µM。然而,与10相比,S连接臂的疏水性对物理化学性质(溶解度 = 0.007 mM,LogP = 3.6,LogD7.4 = 1.1)与选择性(hERG IC50 = 2.8 µM)带来不利影响。尽管 UV/Vis 吸光度仍然很高(Emax 355 nm = 15300 M-1cm-1),但评估光毒性的 3T3 试验仅呈弱阳性(PIF = 5.2;照射IC50 = 16.2 µM)。由于18中的硫醚使苯环移动了大约 1.6 Å 并与10中的邻氯取代基重合,接下来去除了24中的间位氯。单氯取代衍生物25保留了活性(酶学IC50 = 0.070 µM,p-ERK IC50 = 0.250 µM)。与对应的二氯取代衍生物24 (溶解度 = 0.007 mM; LogP = 3.6, LogD7.4 = 1.1)相比,这种结构简化不仅降低了亲脂性 (25: LogP = 2.4, LogD7.4 = 0.8) 而且增加了水溶性 (25: 0.16 mM)。此外,由于在保留活性(例如 25)的情况下允许去除间氯离子,因此尝试用 N 替代间位 C 以及用 CF3 替代邻氯(例如 26)。这不仅保留了活性(26:生化 IC50 = 0.067 µM,p-ERK IC50 = 0.339 µM),同时还降低了亲脂性(26:LogP = 2.0,LogD7.4 = -0.5)并提高了选择性和亲脂配体效率 (hERG IC50 = 17 µM,LipE = 5.0)。事实证明,用氧和碳替换 S-连接臂之后活性变差(27:IC50 = 64 µM,28:IC50 9.9 µM)可能是由于 S 与 ARG111、THR153、LEU254、GLN255 、PRO491口袋的疏水相互作用, 以及亚甲基连接臂朝向出现偏差。不幸的是,尽管与 10 相比化合物 24 的活性有所提高,但由于物理化学性质差、血浆蛋白结合率高无法实现游离型的足够暴露而降低了其在细胞水平的生物活性。
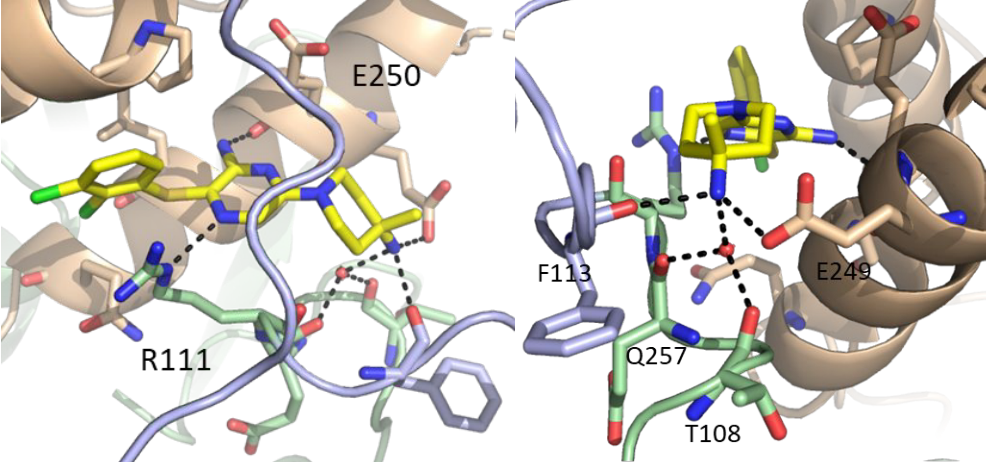
图9. A. 24与SHP2的共晶结构, 1.92 Å, PDB 7JVN, 呈现了氨基吡嗪环与ARG111、GLU250之间的氢键相互作用。B. 24的氨基部分与GLU249、PHE113以及水发生相互作用。图片来源:文献[1]。
24和SHP2的共晶结构(图 9,PDB 7JVN)显示出与之前观察到的10 、18相似的结合相互作用。二氯苯环参与了与ARG111 的阳离子-π堆积相互作用,这可能经由ARG111与吡嗪环N发生氢键相互来预组织完成。与10类似,胺基吡嗪也与GLU250形成氢键。4-甲基哌啶-4-胺片段朝向通道末端的极性残基,与GLU249、PHE113以及结构水形成氢键。由于在生理pH值下的伯胺质子化为正电中心,4-甲基哌啶-4-胺将所有三个质子供给相邻的残基与水(图 9B)。有趣的是,结构水与THR108、GLU110和THR253进一步相互作用。这些远程相互作用更加提起了延伸胺片段以置换结构水并与蛋白质(例如THR108、GLU110或 THR253)直接相互作用的想法。
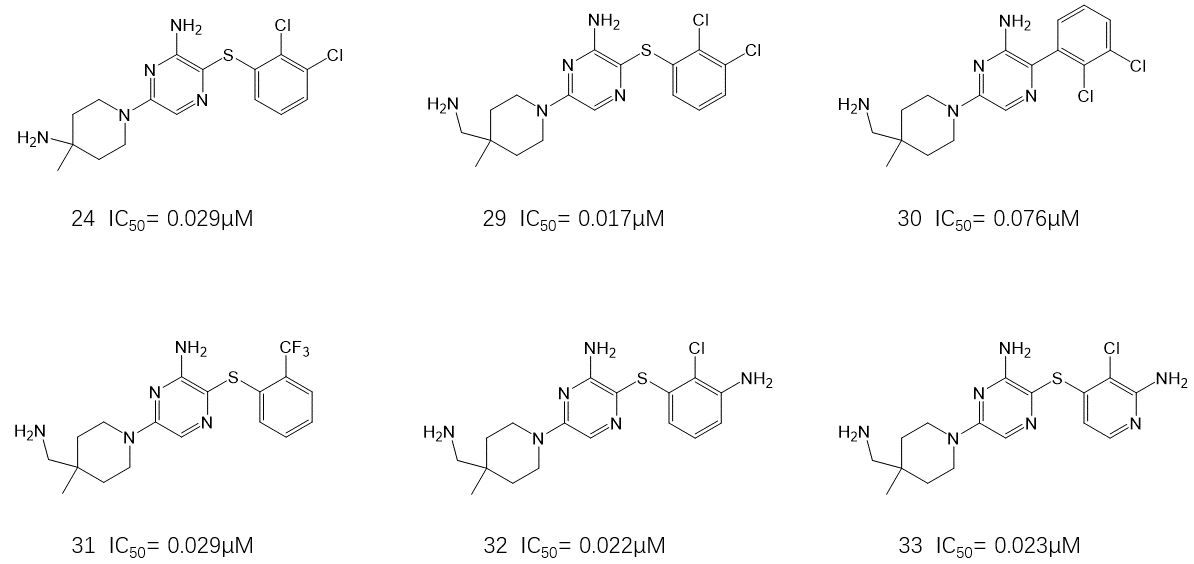
图10. 硫醚芳环的SAR
有了这个想法,作者在24的4-氨基-4-甲基哌啶上插入一个甲基以获得氨基延伸的同系物29(图10:酶学IC50 = 0.017 μM,p-ERK IC50 = 0.088 μM)。这种转化使活性提高了约24倍,并且具有类似的亲脂性(29:LogP = 4.3,LogD7.4 = 2.5)和选择性(29:hERG IC50 = 2.4 μM)。有趣的是,胺基延伸类似物30的活性低于24与10,这表明硫醚连接臂和29延伸的胺之间存在协同作用。如前所述,对间位和邻位取代基(相对于 S)的干扰不影响活性(例如 31、32:IC50 = 0.022-0.029 μM),引入额外的杂原子可以控制亲脂性(31:LogP = 1.0,LogD7.4 = -1.6)。将N原子插入到S的对位,旨在与LYS492(原文为L492,应该为K492)相互作用(见下文),例如33的酶学IC50 = 0.023 μM,p-ERK IC50 = 0.123 μM。与29(LogP=4.3,LogD7.4=2.5,hERG IC50 = 2.4μM)相比, 这不仅降低了疏水性(LogP=1.97,LogD7.4=-0.85),提高了亲脂配体效率(5.2)以及选择性(33:hERG IC50大于30μM)。
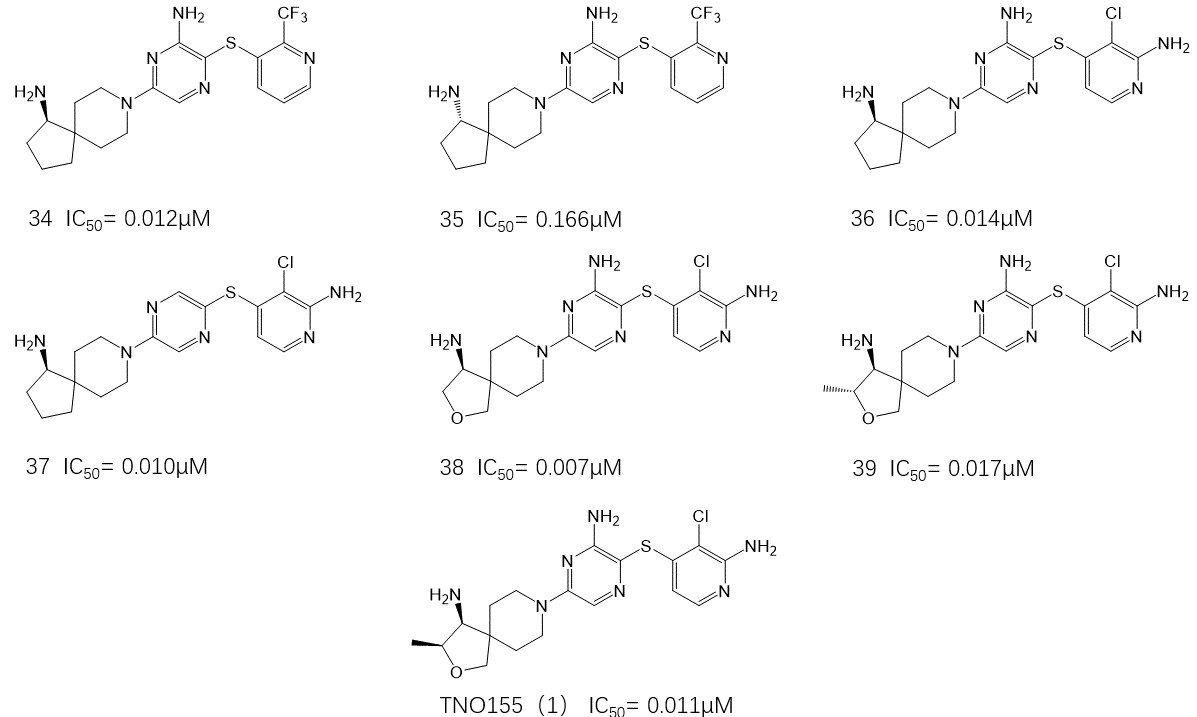
图11. 氨基片段的SAR
硫醚存在下胺的延伸导致活性具有中等程度(2-3 倍)增加,接下来探索如何对延伸的胺进行环化以稳定其构象(图 11)。尽管探索了各种双环(未显示),但 6,5 螺环系统(例如34-38, 1)证明是最有效的。对S和R型胺对映体(例如,化合物 34、35)进行了评估,发现S-胺对映异构体(34:IC50 = 0.012 μM;35:IC50 = 0.166 μM)对活性明显有利。此外,间胺基吡啶衍生物(例如 36)保留了酶学活性并且SAR 与无环系列(如33)没有显着差异,但其细胞水平的活性显着增加(36:p-ERK IC50 = 0.024 μM;33:p-ERK IC50 = 0.123 μM)。值得注意的是,从吡嗪中去除胺基而不丧失结合亲和力、酶学抑制和细胞活性是可能的(例如,37)。该结果表明,随着芳烃和胺基片段的亲和力增加,与早期化合物(例如 10 至 11)相比,与 GLU250 相互作用对于结合的重要性降低。然而,脱氨基的同系物对hERG活性更强且具光毒性。为了控制胺的pKa并避免不利的亲脂性胺相关的泛活性与毒性(例如 hERG、磷脂沉积),在螺环系统中加入额外杂原子(例如 38)并进行评估。虽然与碳衍生物(例如 36:pKa = 9.6)相比,氧气的引入降低了pKa(38:pKa = 7.6),但观察到细胞活性显着丧失(例如,36:p-ERK IC50 = 0.024 μM, 抗增殖 IC50 = 0.123 μM;38:p-ERK IC50 = 0.099 μM,抗增殖 IC50 =0.665 μM) ,但酶学活性没有损失(36:IC50 = 0.014 μM;38 IC50 = 0.007 μM)。为了增加细胞渗透性并平衡整个分子的亲脂性,在呋喃环上增加了一个甲基(例如,39、1)。这保留了酶学活性并提高了细胞活性, S, S-非对映异构体(1,TNO155)具有更优的活性,酶学IC50 = 0.011 μM;p-ERK IC50 =0.011 μM;抗增殖 IC50 = 0.100 μM) 。化合物1溶解度高(0.736mM),具有中等亲脂性(logD7.4=0.6)与高亲脂配体效率(大于6),并且没有测到hERG活性(IC50大于30 μM)。与其它变构通道抑制剂一样,化合物1对磷酸酯酶和激酶组是完全选择性的(见支持信息)。这种选择性是通过配体结合到SHP2通道位点而发生抑制性变构机制来实现。该通道位点为SHP2和SHP1磷酸酯酶所独有的。由于通道位点残基的差异,可能实现对SHP1的选择性。
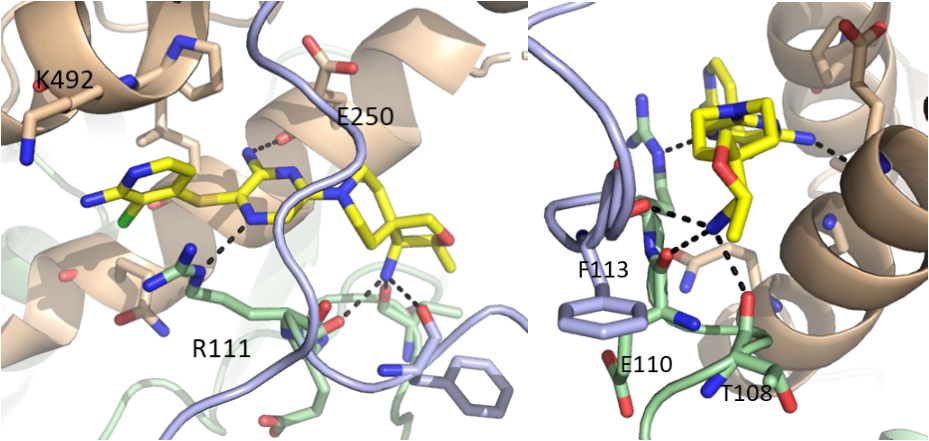
图12. 左:1与SHP2共晶结构, 2.15 Å. PDB code 7JVM。右:氨基片段与SER109, GLU110, PHE113, HIS114的相互作用。图片来源:文献[1]。
1与 SHP2 共晶结构(PDB 7JVM,分辨率为2.15 Å )揭示了几种新的相互作用(图 12)。扩展的胺确实取代了结构水,并与残基 SER109、GLU110 以及PHE113 产生了新的直接相互作用。再一次,ARG111 参与了与氯吡啶环的阳离子-pi堆积相互作用,这是通过与吡嗪 N的氢键相互进行了预组织。如前所述,吡嗪胺与GLU250发生氢键相互作用。 尽管没有观察到正式的 H 键,但LYS492向1的吡啶N移动。吡啶胺官能团与附近的水网络(未显示)相互作用,间接与 LYS492 相互作用。
表1. 化合物1的临床前在多个物种上PK数据
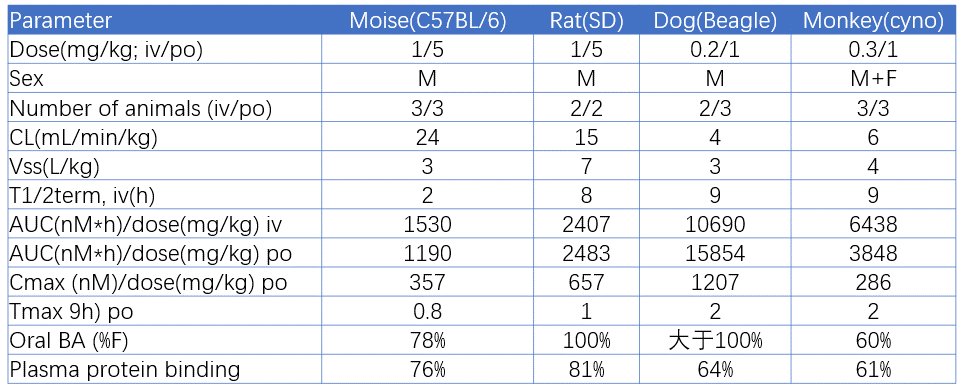
首先在低剂量(0.2 – 1 mg/kg IV,1 – 5 mg/kg PO,悬浮液制剂)下评估1在四种临床前物种(表 1:小鼠、大鼠、狗、猴)上药代动力学特性 。 在所有物种中均观察到中等程度至低的清除率与早期 Tmax(0.8-2.0 小时)以及中等程度的血浆蛋白结合(61-81%)和中等程度到高口服生物利用度(60-100%F)。这些观察结果与有利的理化特性(高溶解性、渗透性)以及在微粒体和肝细胞中低的体外 CL一致。此外,化合物 1 不是 CYP3A4、2D6 或 2C9 抑制剂,因此联合用药时发生药物相互作用的风险得到最小化。
接下来在EGFR驱动的食管癌异种移植瘤模型KYSE-520中对1进行了活性评估。对皮下植入KYSE-520食管癌细胞的小鼠进行了体内PKPD评估(图13 A)。一旦肿瘤达到大约 300 mm3,就单次口服剂量的化合物1并以指定的剂量水平收集血浆和肿瘤组织。分别用qRT-PCR和MSD检测下游PD生物标志物DUSP6(mRNA)和pERK(蛋白)的表达水平。观察到剂量依赖性肿瘤PD药效与1的游离血浆浓度相关。在2周的时间段内多次给药后(图 13B),1显示出强大的剂量依赖性抗肿瘤效果,在10-20 mg/kg BID时达到停滞,不会引起体重减轻。该最大的抗肿瘤效果与之前报道的其他分子(例如 10、厄洛替尼等)一致,但在更低的剂量下实现。重要的是,化合物1在体外3T3 NRU光毒性试验中证明为阴性(PIF = 1.5;照射下的 IC50=666 µM)。
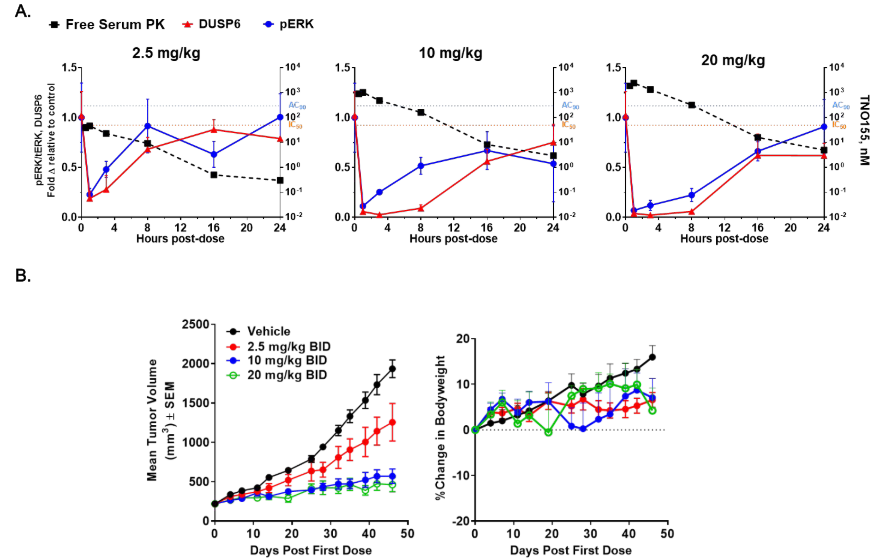
图13. A. 1以2.5、10和10mg/kg剂量口服给药后KYSE-520荷瘤裸鼠中的剂量依赖性暴露、p-ERK和DUSP-6mRNA调节。B. 1在2.5、10和20mg/kg BID剂量下口服给药后KYSE-520荷瘤裸鼠的剂量依赖性药效以及对体重的影响。图片来源:文献[1]。
3. 分析方法
3.1 蛋白的结构准备
将相关复合物晶体结构从蛋白质数据库(PDB)下载到Flare中,并使用来自Protein Prep工具小心地准备,以添加氢原子、优化氢键、消除原子冲突并给蛋白结构分配最佳质子化状态。任何截短的蛋白质链被封端作为蛋白质准备的一部分。使用COBALT多重比对工具在Flare中比对蛋白质序列,随后通过Cα的最小二乘拟合进行叠合。
3.2 GIST分析
了解蛋白活性位点内水分子的行为是药物设计的一个非常重要的方面。在蛋白质-配体复合物中,了解桥连水分子的稳定性是决定药物设计策略的关键。如果水分子不是特别稳定,可以设计配体以取代它,并与蛋白质活性位点直接相互作用:这通常会导致配体生物活性的增加。
GIST是一种水热力学性质分析方法[4],用于评估结合口袋的水合作用,并通过在分子动力学运行结束时对显式溶剂分布采样来计算相关的水热力学性质。GIST分析的结果显示为活性位点水合作用的三维等值面映射“happy”(绿色,与负∆G相关)和“unhappy”(红色,与正∆G相关)区域。unhappy区域映射的是蛋白质活性位点内更可能的可成药区域。happy区域映射的是蛋白结合位点内较低可能成药的区域,在那里水分子更稳定,因此更难置换。
在本文中,用Flare分别对结合位点的Apo结构(不包含配体、金属离子与结晶水)进行了GIST分析,使用了如下条件:
- Calculation method: Normal
- Ligand: None
- Grid spacing:0.5 Å
- Grid Definition:Ligand
- Chains: 不包含水、配体与其它
- Simulation length: 20ns
- Solvent Model: explicit TIP4Pew Water
3.3 蛋白相互作用势分析(PIP)
蛋白相互作用势(Protein Interaction Potential, PIP)是Cresset分子相互作用势对蛋白质的延伸,两者都是使用XED力场计算的。该方法在原理上类似于配体场的计算:蛋白质的活性位点被探针原子充满,并且计算每个格点上的相互作用势。该方法利用了Mehler等人[5]的距离依赖的介电函数来更好地处理蛋白质结构中的大量带电基团。仅计算和显示活性位点的蛋白质相互作用电势。
3.4 静电互补性分析
Flare引入了一种称为静电互补性打分(Electrostatic Complementarity score,EC score)的分析方法[6],它将配体静电的分析与蛋白静电的分析结合起来,以产生配体-蛋白质复合物的静电互补性(EC)的视觉评估和数值评估。基本思想非常简单:当配体和受体的静电势匹配(即具有相同的量值和相反的符号)时,实现配体和受体之间的最大静电亲和力。通过将该方法应用于一系列文献数据集,我们分析了视觉和数值分量,表明它与报道的生物活性差异相关,并能够预测报道的生物活性差异[6]。我们用Flare的EC技术对化合物进行静电互补性比较。
3.5 配体场
Cresset XED力场[7,8]通过复杂的原子描述来模拟远离原子中心的电荷,从而改进了传统的分子力学。这使得能够更详细地描述静电并优异地重现分子间相互作用。XED力场由Andy Vinter博士开发,在Cresset改进,它正确地模拟了取代基对芳香族化合物的效应、复杂芳香族化合物中电荷密度的变化以及小分子、水和蛋白质之间的分子间相互作用。在本文中,我门用XED力场来描述配体的静电性质。
4. 从基于结构的角度理解SAR
4.1 芳环部分的初步SAR
高通量筛选发现苗头化合物2之后,解释了2与SHP2共晶结构PDB 5EHP,并对芳环部分进行了初步SAR探讨。这里,我们用经典的AutoDock Vina[2]进行分子对接打分,以考察打分函数能否区分2、3、4、5的活性差异,结果如表2所示。发现四个化合物的分子对接打分值几乎一样,没有活性的5甚至打分最佳。这说明,Vina不能描述苯环邻、间位双氯取代的重要性,也不能区分氯与甲基的差异。
表2. 化合物1、2的AutoDock Vina(Version 1.2)分子对接打分结果
| Comp ID | PDB ID | Vina Score(kcal/mol) | Hydrophobic | H-Bond |
|---|---|---|---|---|
| 2 | 5EHP | -10.25 | 33.74 | 0.64 |
| 3 | 5EHP | -9.83 | 32.82 | 0.70 |
| 4 | 5EHP | -9.90 | 32.24 | 0.70 |
| 5 | 5EHP | -10.63 | 43.83 | 0.69 |
采用Roche公司Kohn等人[3]的非经典相互作用分析方法,可以发现邻、间-位氯涉及多种高频的相互作用。
图14分析了邻位氯的相互作用。可以发现Cl与ARG111侧链CG、CD碳、THR253侧链GC2碳以及GLN257侧链GC碳共发生4次Cl…ali_apol模式(RF=1.13)的疏水相互作用。与THR253主链羰基氧与碳分别发生1次卤键以及1次Cl…C_pi_carbonyl(RF=2.18)相互作用。与LEU254主链酰胺N发生1次Cl…N_pi_don模式(RF=1.51)相互作用,与LEU254的CA发生1次Cl…C_ali_don(RF=1.57)模式的弱极性相互作用。

图14. 化合物2与SHP2共晶结构PDB 5EHP结合位点中,化合物2芳基片段邻位氯的非经典相互作用。
图15分显示了间位氯的非经典相互作用。可以发现Cl与GLN495侧链的CA、CB,LEU254侧链的CD1,GLN257侧链的CB、CG等共发生了5次疏水的Cl…ali_apol模式(RF=1.13)相互作用;Cl与GLN495的酰胺片段发生3次弱极性相互作用,分别为Cl…N_pi_don(RF=1.51)、Cl…C_pi_carbonyl(RF=2.18)以及Cl…O_pi_acc(RF=1.45)。
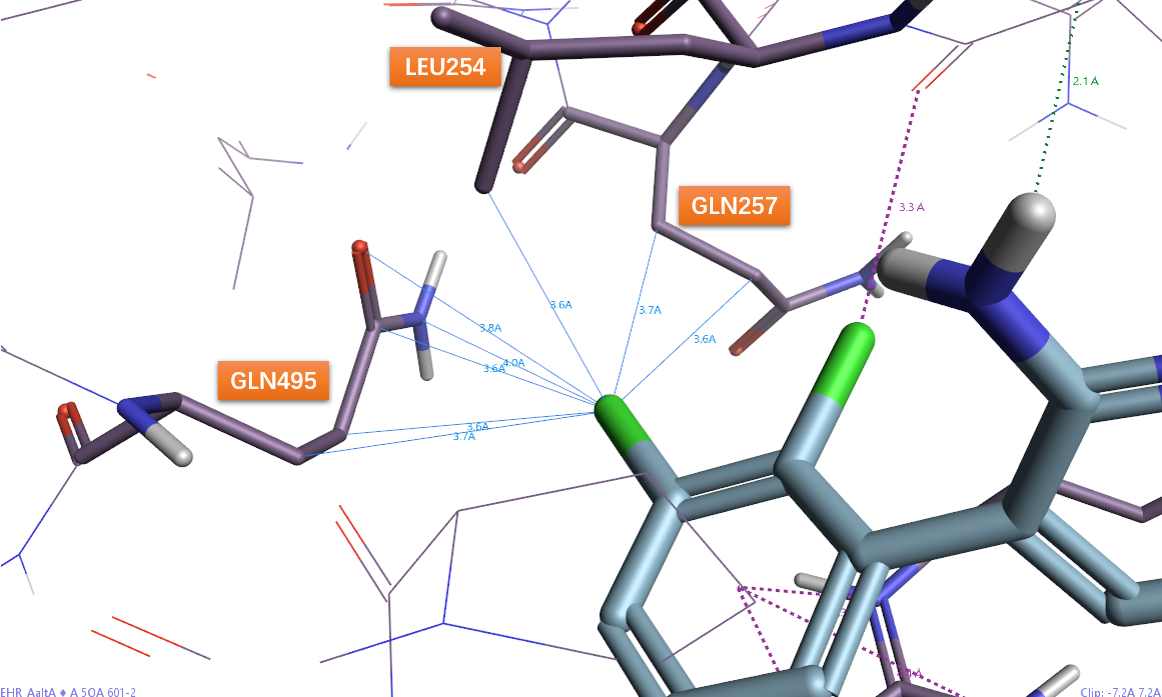
图15. 化合物2与SHP2共晶结构PDB 5EHP结合位点中,化合物2芳基片段间位氯的非经典相互作用。
用Flare编辑PDB 5EHP配体化合物2芳基片段的对位,替换氢为氯,并对氯用XED力场进行优化后开始分析相互作用,结果如图16所示。对位氯分别与GLN495侧链的CG、CB碳原子,LYS492侧链的CB碳原子发生3次的Cl…ali_apol(RF=1.13)模式的疏水相互作用。对位氯与LYS492主链CA原子之间发生生1次Cl…C_ali_don(RF=1.57)模式的极性相互作用。图16中还绘出与Cl接触的几个酰胺片段原子,因为角度不满足要求而没有计入相互作用。
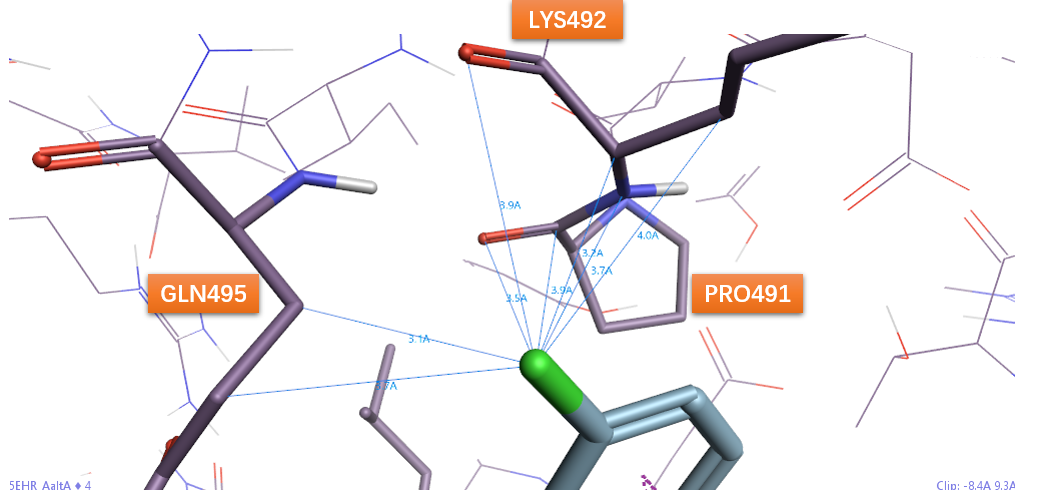
图16. 化合物9(在PDB 5EHP结合位点里对化合物2进行编辑而来)芳基片段对位氯与SHP2的非经典相互作用。
比较化合物2、3、4、5极性相互作用数量,从多到少依次为2、4、3、5,这与化合物的其活性强弱顺序一致。尤其是化合物2的邻位氯发生了作用很强的卤键相互作用,因此活性最高,而失去邻位氯之后的3活性几乎丧失,这充分证明了邻位氯的重要性。在3的对位引入氯之后的4,活性几乎增强了1倍,但弱于2,这说明对位氯的重要性低于邻位。总的来说,氯看起来不只是单纯的通过疏水性与蛋白相互作用,其极性相互作用的数量与活性直接相关。
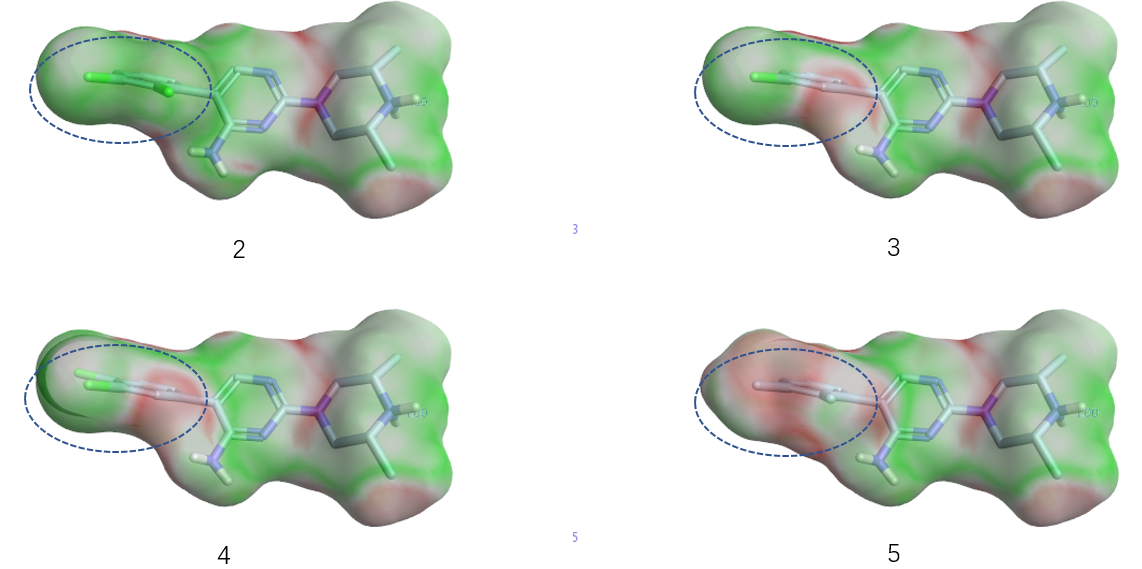
图17. 化合物2、3、4、5与5EHP的表面静电互补性分析。虚线圆圈:芳环所处的表面。绿色:静电互补;红色:静电冲突。
在PDB 5EHP结合位点里计算化合物2、3、4、5与SHP2之间的静电互补性,结果入图17所示。化合物2的芳环表面基本为绿色,3、4的芳环部分红色部分绿色、而5的芳环表面基本为红色,这说明化合物2与蛋白之间具有最好的静电互补性,而3、4、5在静电上与蛋白具有冲突(5冲突最严重),这与化合物2的活性最佳、3、4次之、5最次的事实一致。结果表明,表面静电互补性程度可以预见配体与蛋白在静电上不匹配的位置、解释化合物芳环部分的SAR,并可依此设计新的化合物。
用Flare GIST分析化合物2与SHP2共晶结构PDB 5EHP的Apo结合位点,结果如图18所示。可以发现,邻、间位氯被ΔG=+0.5kcal/mol等值图(图18 左)标号为W1、W2区块覆盖,说明这两个氯从去溶剂化自由能获益。
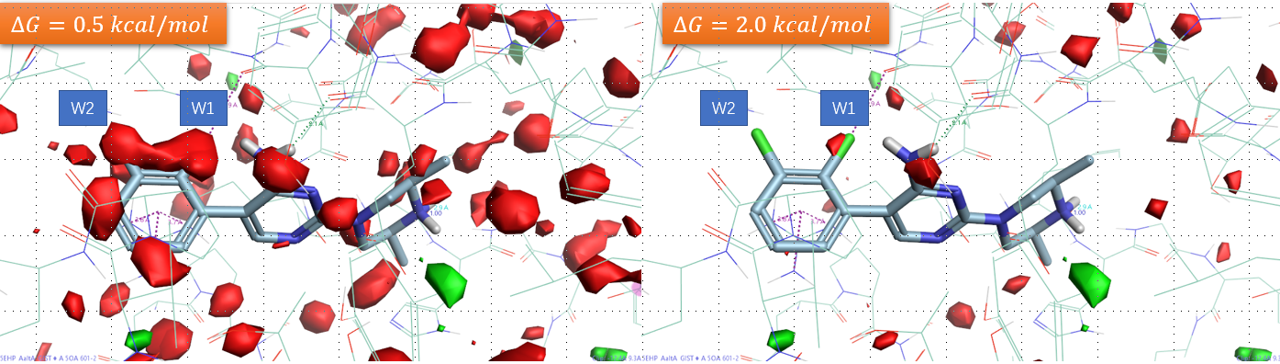
图18. 共晶结构5EHP结合位点Apo结构的GIST分析。左红色等值图:ΔG=+0.5kcal/mol; 右红色等值图:ΔG=+2.0kcal/mol。
为了突出显示重要的溶剂化位点,提高等值图至ΔG=+2.0kcal/mol(图18右),可以发现W2(间位氯)等值图消失,而W1(邻位氯)等值图还存在,这说明邻位氯比间位更重要。将GIST的溶剂化ΔG等值图与蛋白相互作用势、静电互补性分析相结合,比较容易做出保留邻、间位氯的决定。
4.2 哌嗪环的初步SAR
同样的,将上一步GIST分析结果与蛋白相互作用势一起观察有助于分析哌嗪环的SAR。图19展示了5EHP复合物结合位点Apo结构的GIST分析结果(红色等值图:ΔG=+0.5kcal/mol)以及蛋白相互作用势(蓝色透明等值图:负蛋白相互作用势;深红色透明等值图:正蛋白相互作用势)。可以看到化合物2(图19左的配体)哌嗪环阳离子中心氮平伏键取向上有两个挨着的个红色ΔG=+0.5kcal/mol区块(图19 W3与W4),并且位于蓝色透明的负蛋白相互作用势区块里。这意味将正电中心氨基往W3,W4移动(即延长氨基)可能会因为对不稳定水的置换(不影响静电相互作用情况下)而获益。

图19. 共晶结构5EHP结合位点Apo结构的GIST分析。红色实心等值图:GIST ΔG=+0.5kcal/mol; 蓝色透明等值图:负蛋白相互作用势;深红色透明等值图:正蛋白相互作用势。配体(左):化合物2;配体(右):化合物10。
将PDB 5EHR按Cα最小二乘拟合叠合到PDB 5EHP,在5EHP的结合位点里比较5EHR共晶配体10与5EHP的GIST等值图(图19右),可以发现化合物10的哌啶环平伏键上的阳离子中心与W4重合,也就是质子化的氨基替换了不稳定的水W4,同时与THR108、GLU110、PHE113的羰基氧发生氢键相互作用、与GLU249末端羧基产生盐桥相互作用。调整GIST等制图直到ΔG=3kcal/mol(未显示),W4依旧存在,这说明对W4水替换的重要性。
对化合物10与SHP2共晶结构PDB 5EHR的Apo结合位点进行GIST分析也得到相似的结果,如图20所示(为了清晰起见,将蛋白结构隐藏,),从两个视角展示了10与GIST ΔG=+0.5kcal/mol水平等值图、蛋白相互作用势。可以发现,化合物10哌啶环4位的氨基确实与W3、W4同时叠合,并与周围负的蛋白相互作用势互补。
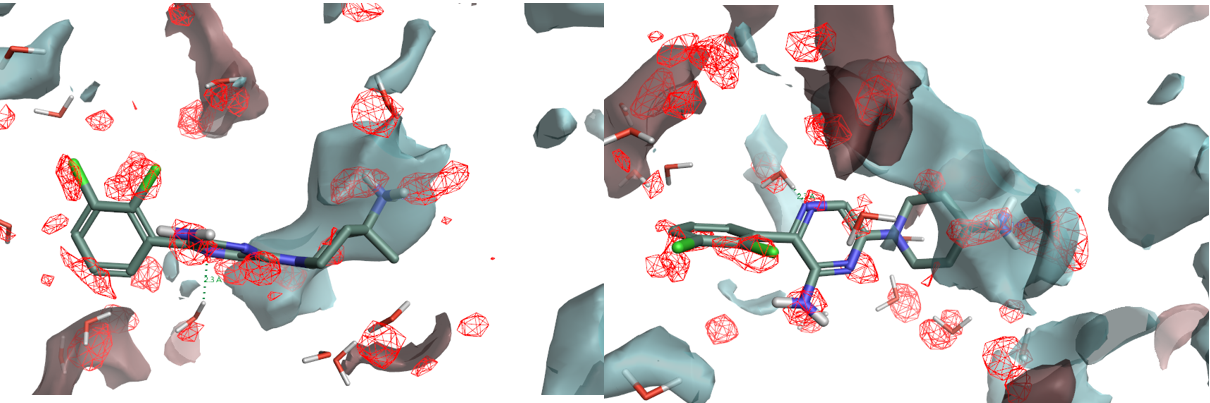
图20. 共晶结构5EHR结合位点Apo结构的GIST分析。红色实心等值图:GIST ΔG=+0.5kcal/mol; 蓝色透明等值图:负蛋白相互作用势;深红色透明等值图:正蛋白相互作用势。配体:化合物10。
总的来说,结合蛋白相互作用势与GIST分析,药化专家在这些明确、清晰的可视化信息的支持下,相信可以构思出延伸哌嗪质子化氮为4-氨基哌啶的方案,实现从化合物2到化合物6的优化(活性提升10倍),甚至到化合物8的优化(活性提升46倍)。如果借助SPARK对W4水进行替换,可以更快的实现胺基片段的优化,具体见第5小节。
4.3 杂环母核的初步SAR
用Flare分析5EHP结合位点的蛋白相互作用势,结果如图21所示,可以发现苗头化合物2杂环母核的嘧啶在静电上与蛋白存在冲突。主要体现在嘧啶环的N-2(编号见图5-9)朝向蛋白蓝色的负静电势区;而N-2邻位的CH指向蛋白的正静电势区,这与配体场(图22 左)的正配体场冲突,对活性不利。
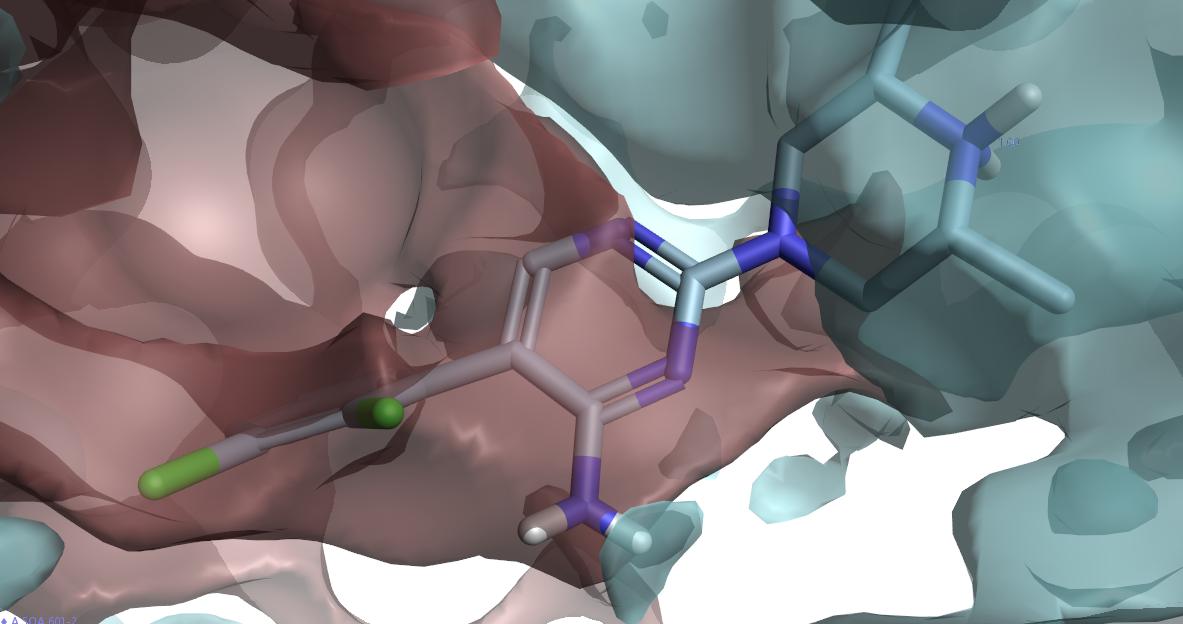
图21. PDB 5EHP结合位点的蛋白相互作用势,主要展示与配体杂环母核对应的部分
从化合物2的配体场(图22左)可以发现,嘧啶N-2被负静电区块包围,而其邻位CH被正静电区块覆盖,这与图21的蛋白相互作用势冲突。
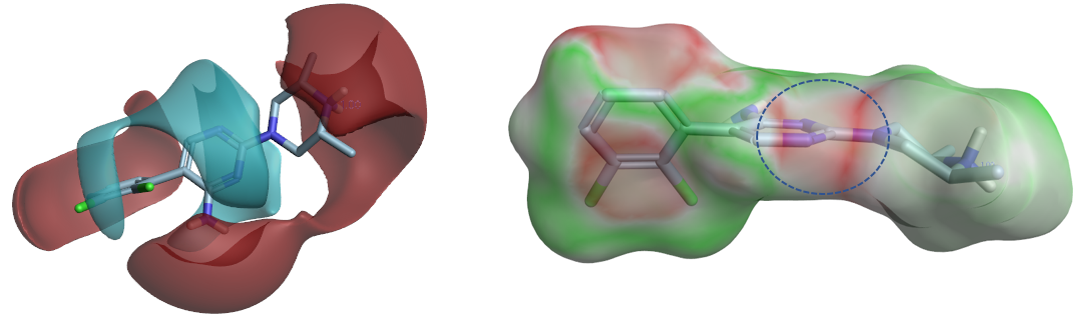
图22. 共晶结构5EHP中化合物2的配体场(左)与静电互补性分析(右)
图22右展示了化合物2的表面静电互补性,可以清晰地发现嘧啶N-2区域被着色为静电冲突的红色(见图22圆圈高亮展示部分),这强烈提示要移除N-2原子以修复静电冲突。
对5EHP结合位点的蛋白相互作用场分析、配体场分析以及表面静电势分析显而易见地让我门联想到将N-2移动到N-1(编号见图5-9)位置的想法:因为这样就可以完美的解决静电冲突。对杂环母核的SAR研究证明了这一点,发现氨基吡嗪母核具有最优的活性(见图5)。比之化合物6,N-2移动到N-1的化合物8活性提升了3.7倍。
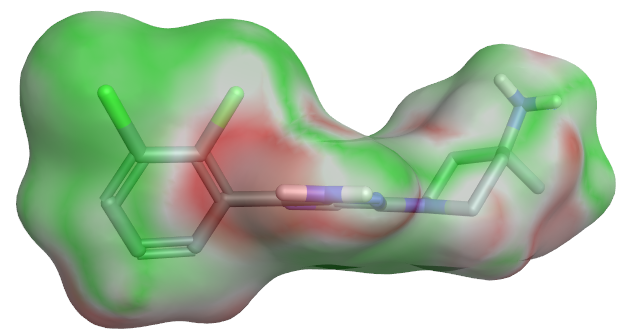
图23. 化合物10的表面静电互补性分析。红色:静电冲突区; 绿色:静电互补区。
观察图18的蛋白相互作用势,可以发现杂环上的氨基与蛋白相互作用势匹配的相当好:其中一个氢指向一个蓝色的负静电势区域;另一个氢稍不如意,其氢原子从红色的正静电势区中突出来。进一步的静电互补性分析(图23)可以发现,母核杂环氨基确实有部分区域处于静电冲突状态。与10相比,移除了氨基的11活性降低了81倍;用甲基取代氨基的一个极性氢后的化合物12的活性降低了314倍。
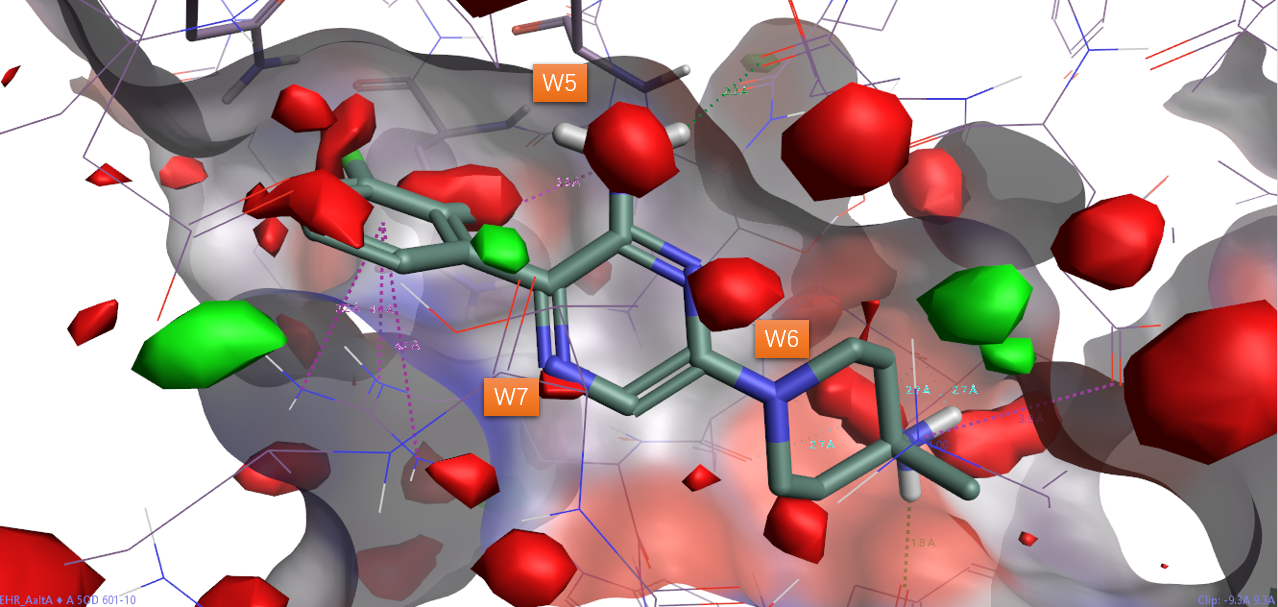
图24. 5EHR结合位点Apo结构GIST分析结果。红色等值图:ΔG=+0.5kcal/mol; 绿色等值图:ΔG=-0.5kcal/mol。
对5EHR结合位点Apo结构的GIST分析还发现,如图24所示,化合物10氨基吡嗪母核的杂原子N以及氨基与GIST的三个高能水W5、W6、W7非常巧合的重合在一起。如果将等值图ΔG提高到1kcal/mol(图25,粉红色)与3kcal/mol(图25,青灰色)以凸显更加重要的高能水合位点,可以发现氨基与最高能的W5重合,氨基邻位的吡嗪氮与次高能的W6重合;氨基对位吡嗪氮与能量最低的W7重合。W5、W6、W7三个水合位点重要性与化合物SAR直接相关:氮与越重要的水合位点重合对活性有利,水合位点能量越高对应的氮越重要。比如,12比10少了个与W5重合的氨基,其活性降低了81倍,当然如前所述,这里面也离不开静电的原因。

图25. 5EHR结合位点Apo结构GIST分析结果。粉红色等值图:ΔG=+1.0kcal/mol; 青灰色等值图:ΔG=3.0kcal/mol。
4.4 硫连接臂引入后相关的SAR
比较化合物10与24的SHP2共晶结构结合位点,如图26所示,可以发现结合位点里的残基ARG111侧链的胍基取向发生了重大变化。在与化合物10结合时,胍基与10的芳环部分基本平行,发生了阳离子-π相互作用,10吡嗪环上的氮在胍基平面外;在与化合物24结合时,胍基发生扭转,与24的二氯苯基不再平行,24的吡嗪氮与胍基共平面,使得胍基与N之间的氢键相互作用角度更加适宜。

图26. 化合物10(PDB 5EHR)与24(PDB 7JVN)结合模式的比较。配体(左):24;配体(右):10
由于受到插入硫连接臂的影响,化合物24的二氯苯基比之10的二氯苯基发生了偏移,氯原子的相互作用模式也发生了变化。具体的,24的邻位氯远离了THR253,不再与羰基氧发生卤键相互作用。邻位氯发生的作用模式(见图27)有:(1)与GLN257、GLN495发生四次的疏水Cl…ali_apol(RF=1.13)相互作用;(2)与GLN495发生1次Cl…C_pi_carbonyl相互作用。间位氯更多暴露在溶剂中,主要与GLN495发生两次的疏水Cl…ali_apol(RF=1.13)相互作用,此外还替换了出现在5EHR结合位点中的HOH1035(见图26右)。间位氯在后续的优化中被更亲水、更适宜于暴露于溶剂中的氨基替换(比如化合物1),也可认为氨基是对HOH1035更适宜的模拟。
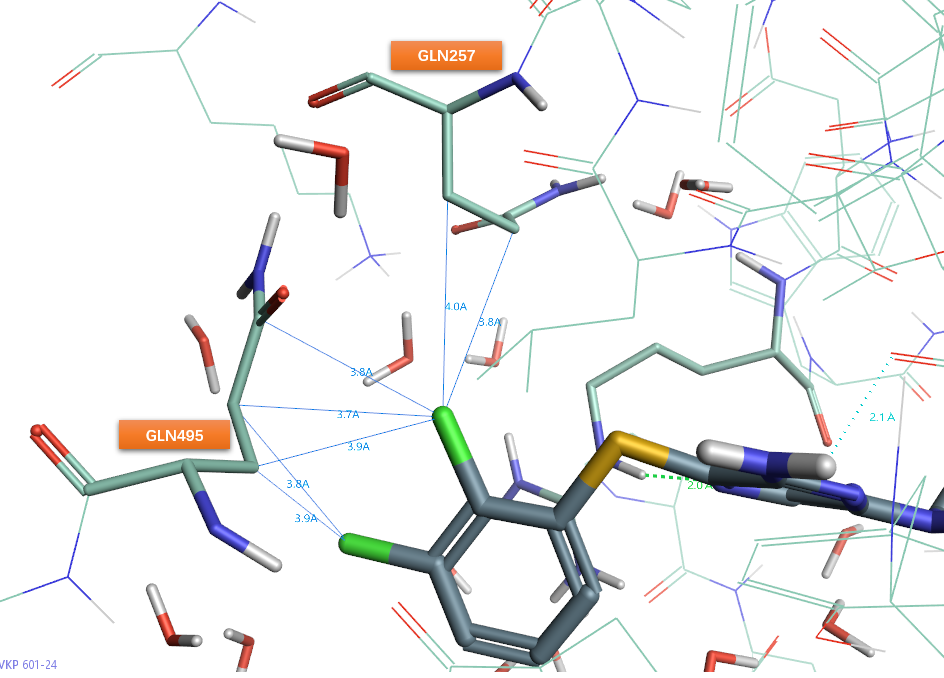
图27. 化合物24邻、间位氯的相互作用
虽然之前的分析显示10、24的二氯苯环在相互作用模式上有所差异,但根据表面静电互补性分析结果(图28)没有发现两者二氯苯基的表面静电互补性有很大的差异。两者都在苯环4、5位方向上都出现了静电冲突。这也提示了苯环4、5位是可继续优化的。在后续的优化中,苯环的4位碳被氮替换,并使得LYS492伸向溶剂的侧链折回与之发生氢键相互作用。
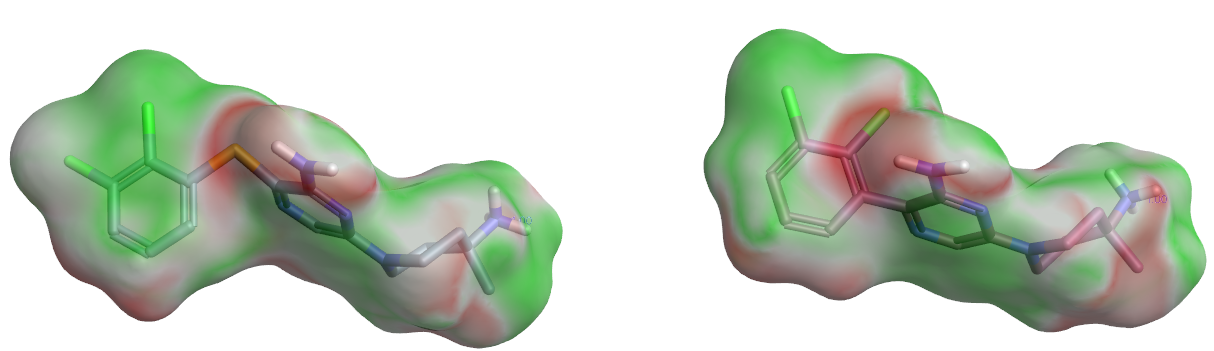
图28. 化合物10、24的表面静电互补性分析
在4.3小节分析哌嗪环的SAR时,认为10的哌啶氨基替换了高能的W4水(见图19、20)而获得结合能上的增益。如图29所示,在7JVN化合物24的结合位点中,在W4对应的位置上出现了一个真实的结合水HOH712(见图29),也就是说计算的W4被证实真实存在!一方面说明GIST算法的正确性,以及之前假设化合物10的4-氨基哌啶是通过替换W4而获得结合能增益是对的;另一方面也说明了24的氨基有别于10的氨基:24的氨基是通过与HOH712相互作用来稳定配体与蛋白的结合,而10是通过对水的替换(模拟)。要注意,如附图2所示,10的胺基并不与水完全重合(水712的O与10胺基N之间的距离为1.8Å,小于水分子的半径,因此认为重合),因此胺基与水所相互作用的残基也不是完全相同,胺基不能算是完全对水模拟。
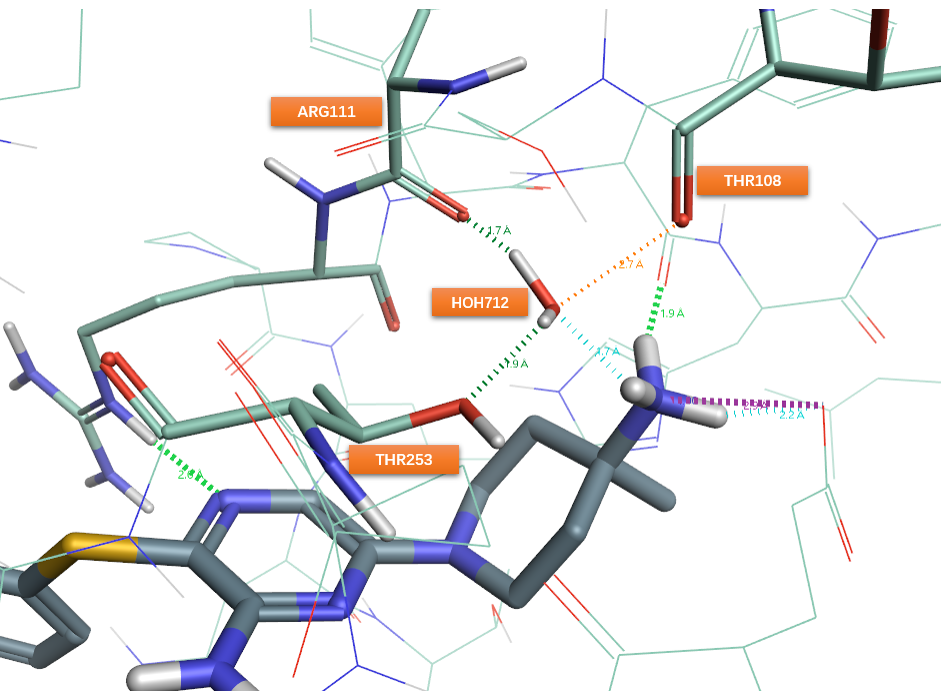
图29. PDB 7JVN结合位点HOH712的氢键网络
由于HOH712直接与THR108、GLU110以及THR253发生氢键相互作用,HOH712还与配体24哌啶4位上的氨基发生氢键相互作用,这使得HOH712是桥连这些氢键网络的中心(见图29),这很容易让人产生一个想法:延伸化合物24哌啶环上的氨基以替换HOH712,可以模拟水与蛋白质THR108、GLU110、THR253的直接相互作用。
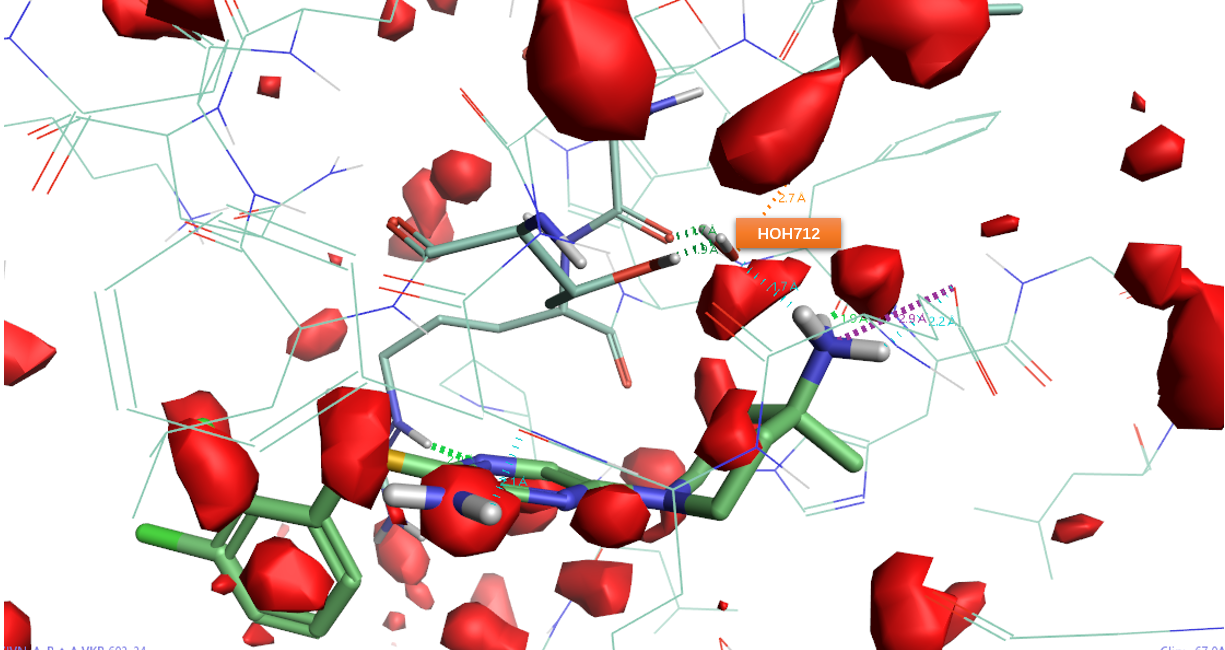
图30. 化合物24与SHP2共晶结构PDB 7JVN结合位点的GIST分析。红色区块:ΔG=+0.5kcal/mol等值图。
用Flare对化合物24与SHP2共晶结构PDB 7JVN结合位点进行GIST分析,结果如图30所示,可以发现化HOH712确实被一个红色区块的“unhappy water”覆盖。实际上,HOH712的ΔG大于3kcal/mol,这足以提示替换HOH712将获益匪浅。具体的如何利用结合水的信息进行结构优化,请参阅第5节。
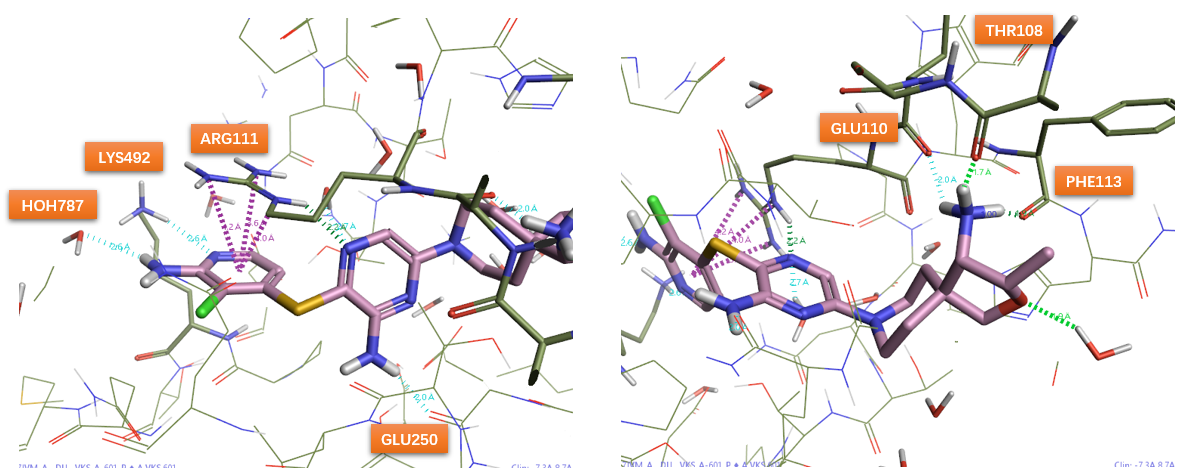
图31. PDB 7JVM结合位点的相互作用
最终进入临床研究的候选化合物1是4-氨基哌啶环延伸的代表,从其与SHP2的共晶结构可以发现,延伸后的氨基刚好代替了HOH712直接与THR108,GLU110、PHE113发生氢键相互作用,见图31右。而芳基部分一方面与ARG111发生阳离子-π相互作用,对位由于引入了碱性的N与LYS492发生氢键相互作用;氨基置换了HOH1035并暴露于溶剂中;氯原子与GLN495、LEU254、GLN257有5次的Cl…ali_apol疏水作用(RF=1.13);与GLU495有1次的1次Cl…C_pi_carbonyl(RF=2.18)相互作用。吡嗪环的N与ARG111的胍基共平面,这有利于胍基与吡嗪的氢键相互作用。此外吡嗪环部分的氨基作为供体与GLU250发生氢键相互作用。总的来说,化合物1集成了各个子片段各方面优势。
5. 利用SAR信息加速药物发现:胺基部分的水分子替换
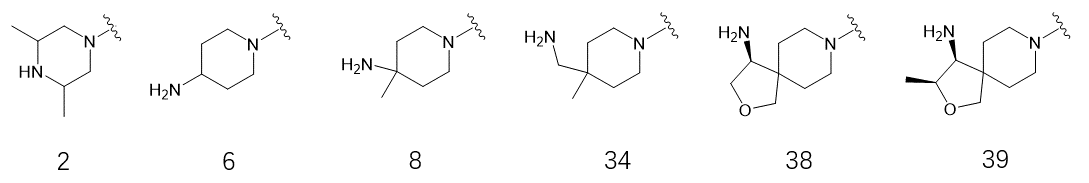
图32. 胺基部分的发展过程:仅列出对活性有利的胺基片段形式,按第一次出现的分子序号排序
图32显示了胺基部分的发展过程,列出对活性有利的胺基片段形式(按第一次出现的分子序号排序)。胺基优化过程实际是与结构生物学直接相关的,经历了三个阶段,有三个不同逻辑:
- 苗头化合物2的共晶结构PDB 5EHP的解释:延伸残基,引入新相互作用
- 先导化合物24共晶结构PDB 7JVN的解释:替换结合水HOH712,引入新相互作用
- 优化阶段
根据苗头化合物2的共晶结构,2的哌嗪环片段占据了别构结合位点内由PHE113、HIS114、GLU249、GLU250、THR218等残基组成的极性腔,同时也暴露于溶剂中。假设将胺基往该这些极性残基延申使得发生新的相互作用成为可能,并增加对磷酸酯酶的抑制活性。该阶段代表性胺片段如6、8。
在发现7JVN的结合水HOH712介导这几个残基的相互后,尝试在氨基与哌啶之间插入一个亚甲基以延长氨基实现HOH712的替换,使得分子直接与蛋白发生新的相互作用。延伸之后的代表性胺基片段如34。
接下来探索如何稳定胺的构象同时改善DMPK与hERG毒性,其结果是设计了螺环类化合物,代表性胺基片段如38、39。
实际上,这个优化过程可以得到加速。如前面4.2小节所述,根据化合物2共晶结构PDB 5EHP的GIST分析,就已经预测到HOH712的存在(即图19的W4),并在对化合物10共晶结构PDB 5EHR的GIST分析中进一步确认。根据预测水进行水分子替换,我们之前已经在《借助重要的水加速基于结构的ATX抑制剂先导化合物优化》一文中详细地演示过如何用SPARK实现。本文为了简化,以化合物24为起点,利用PDB 7JVN的信息演示水分子替换的效果。
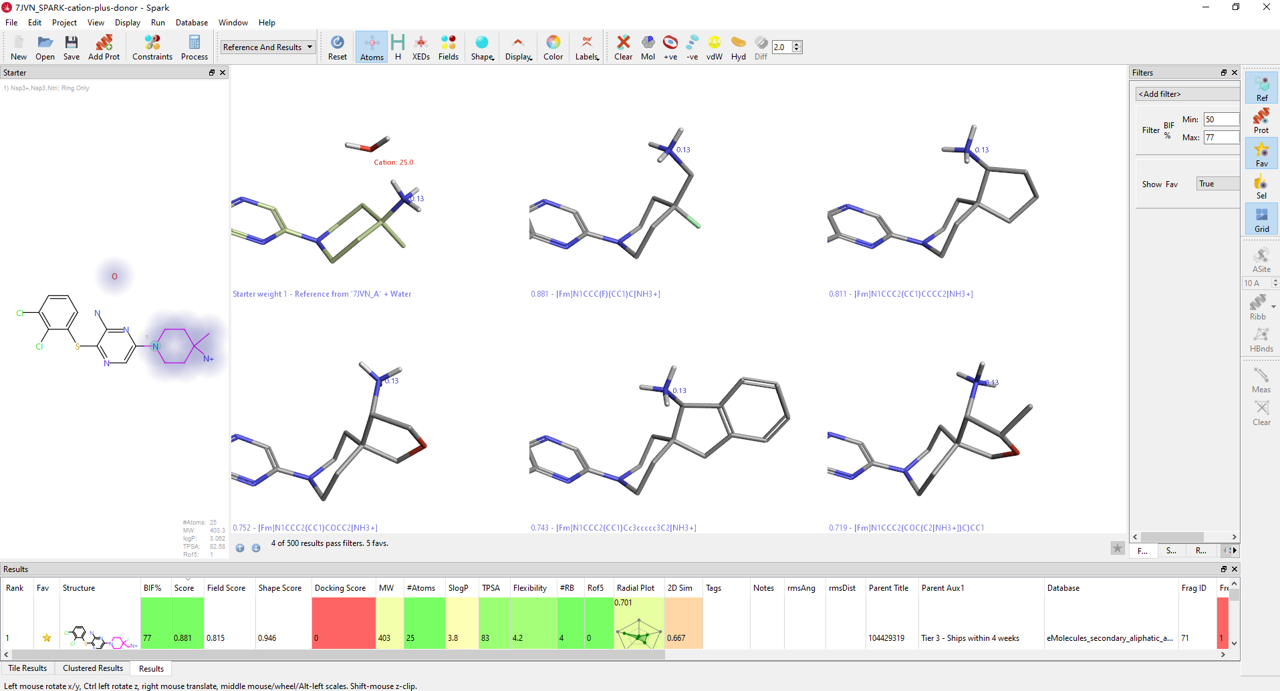
图33. 以PDB 7JVN的共晶化合物24为起点,借助SPARK对哌啶与HOH712进行水分子替换计算实验的结果。
在本次计算实验中,以PDB 7JVN的共晶化合物24为起点(见图33),借助SPARK对哌啶与HOH712进行水分子替换计算实验(见图33),给出500个结构(详细内容,请下载附件1)。根据SPARK打分的排序,图32中代表性胺34、38、39的排名分别为1、41、77(见图34),代表性的胺基延伸类型全部被搜索命中。
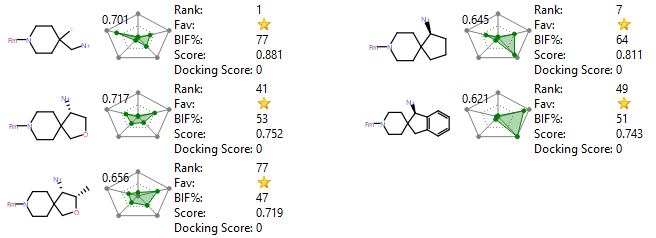
图34. SPARK命中的典型结构类型及其打分排名
此外,除了命中文章中公开的胺类型之外,还命中了两个相关结构,即打分排名第7与49的全碳类型的螺环(见图35),这两个片段在诸多的SHP2抑制剂专利申请里经常出现,比如北京加思科的CN201980063878.0、上海拓界的CN202011047026.3、与南京圣和的CN202011228125.1等中国发明专利申请就公开了这两个片段。
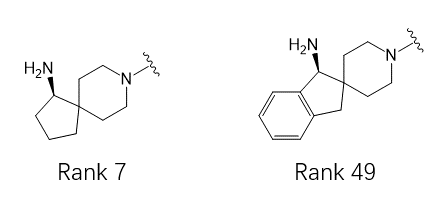
图35. SPARK打分排名第7与49的结构。
总的来说,借助SPARK的水分子替换实验,可以实现胺的延伸并替换结合水HOH712的目的,最终实现加速先导化合物的优化。如果利用预测的水分子进行水分子替换实验,可以将这个优化进一步提前到苗头化合物2复合物5EHP解释出来的时候。
6. 硫醚连接臂的SAR
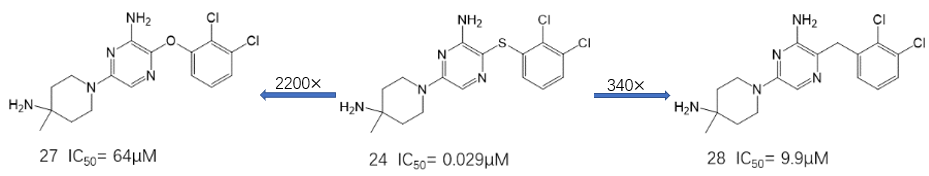
图36. 硫醚连接臂的SAR
在探索硫醚片段连接臂SAR的时候,除了硫连接臂(比如24,图36)之外,还测试了氧与碳连接臂(比如27与28,图36)。出乎意料地,比之硫连接臂24(IC50=0.029μM),氧连接臂27(IC50=64μM)与碳连接臂28(IC50=9.9μM)的活性分别降低了2200与340倍。然而,基于24与SHP2共晶结构PDB 2JVN的分析,却发现不了三者在相互作用模式上的差异,也没现在去溶剂化上的差异。用经典的分子对接软件AutoDock Vina对这三个化合物进行打分,结果如表3所示。在Vina打分函数中,三者的氢键贡献一致,而氧、碳连接臂由于疏水贡献更大,从数值上看其结合自由能略优于硫连接臂,这与实验并不一致。分子对接Vina的打分解释不了活性差异。
表3. 化合物24、27与28的Vina分子对接打分值比较
| Comp ID | PDB ID | Vina Score(kcal/mol) | Hydrophobic | H-Bond |
|---|---|---|---|---|
| 24 | 7JVN | -9.80 | 21.24 | 3.73 |
| 27 | 7JVN | -10.85 | 21.15 | 3.73 |
| 28 | 7JVN | -11.11 | 30.48 | 3.73 |
在一般情况下,如果从相互作用与去溶剂化自由能角度不能解释活性差异时候,配体张力能可能是活性差异的原因。为了简化计算,仅对3个化合物的连接臂片段部分进行计算,三个连接臂片段结构如图36所示。
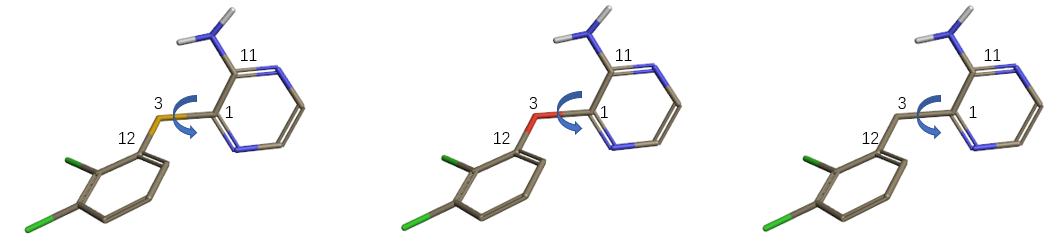
图36. 硫、氧、碳三种连接臂片段
计算过程如图37所示,首先用Flare的Conf Hunt对化合物进行构象搜索,XED力场几何优化,得到初始的构象系综;然后用Flare QM的半经验方法xTB-GFN2对构象系综进行几何优化,并进行构象聚类,去掉重复的构象(RMSD=0.125埃 并且 ΔE\( \gt \)0.01kcal/mol),得到新的构象系综;然后在BP86-D3BJ/DEF2-TZVP理论水平对新的构象系综进行几何优化、找到全局最低能构象;以全局最低能构象为参比,在wB97X-D/DEF2-TZVP理论水平计算生物活性构象的相对张力能。
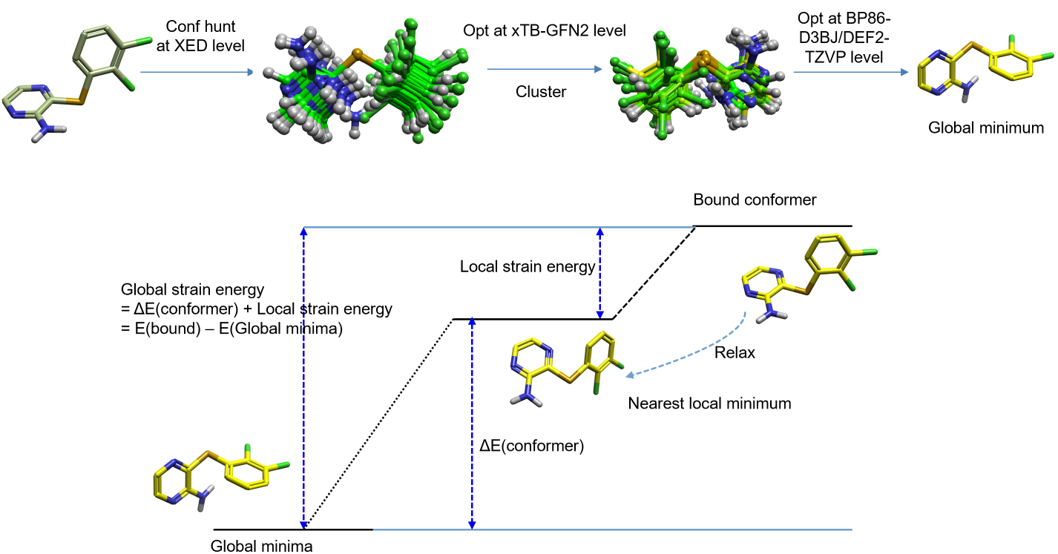
图37. 硫、氧、碳三种连接臂片段的活性构象张力能计算方法
化合物24、27、28连接臂片段的构象张力能计算结果如表4所示。可以发现,比之碳、氧链接臂,硫链接臂具有最低的全局构象张力能,硫连接臂化合物24也是三个里活性最强的;但是,计算的碳、氧连接臂构象张力能与其结合自由能的排序并不一致。
表4. 三个链接臂的生物活性构象张力能计算结果
| Items | 硫/24 | 氧/27 | 碳/28 |
|---|---|---|---|
| EBound(Hartree) | -1868.216671 | -1545.233234 | -1509.325646 |
| Elocal minimum(Hartree) | -1868.218741 | -1545.233233 | -1509.325647 |
| EGlobal minimum(Hartree) | -1868.218845 | -1545.236619 | -1509.331517 |
| Global ΔE(kcal/mol) | 1.36 | 2.12 | 3.68 |
| ΔGa (kcal/mol) | -10.27 | -5.72 | -6.82 |
a.根据IC50转化而来
还可以发现,C、O连接臂的生物活性构象就是局部极小点,因此它们的张力能主要来自ΔE(conformer);而S连接臂的生物活性构象与最近的局部极小点发生两面角扭曲,但是局部极小点几乎接近全局最小点,因此硫链接臂的张力能主要来自诱导契合导致的配体构象焓变。
7. 附件
- PDB 5JVN结合水HOH712的水分子替换实验
- 5EHR与7JVN叠合后的共晶配体10、24以及结合水HOH712的位置比较
水分子替换实验的SPARK项目文件:7JVN_HOH712_water_replace.fsp。
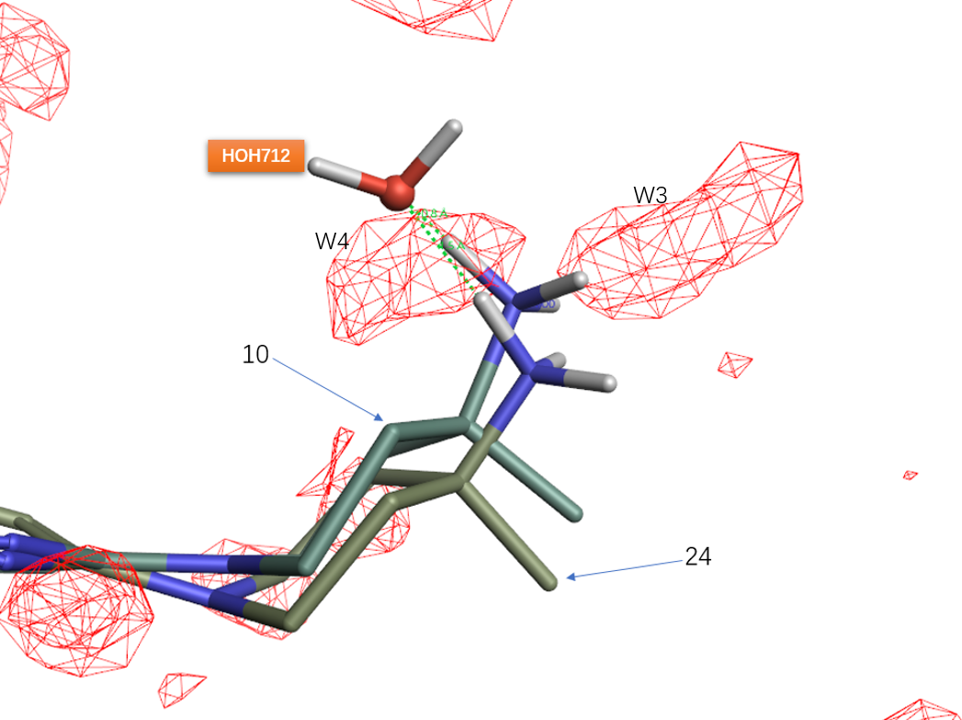
将7JVN按Cα叠合到5EHR上,比较共晶的配体24、10以及7JVN的HOH712位置关系,其中网格状等值图是对5EHR的Apo结构进行GIST分析得到的 ΔG=+0.5kcal/mol等值图。
8. 文献
- Lamarche, M. J.; Acker, M.; Argintaru, A.; Bauer, D.; Boisclair, J.; Chan, H.; Chen, C. H. T.; Chen, Y. N.; Chen, Z.; Deng, Z.; et al. Identification of TNO155, an Allosteric SHP2 Inhibitor for the Treatment of Cancer. J. Med. Chem. 2020, 63 (22), 13578–13594. https://doi.org/10.1021/acs.jmedchem.0c01170.
- Eberhardt, J.; Santos-Martins, D.; Tillack, A. F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, acs.jcim.1c00203. https://doi.org/10.1021/acs.jcim.1c00203.
- Kuhn, B.; Gilberg, E.; Taylor, R.; Cole, J.; Korb, O. How Significant Are Unusual Protein–Ligand Interactions? Insights from Database Mining. J. Med. Chem. 2019, 62 (22), 10441–10455. https://doi.org/10.1021/acs.jmedchem.9b01545.
- Nguyen, C.; Gilson, M. K.; Young, T. Structure and Thermodynamics of Molecular Hydration via Grid Inhomogeneous Solvation Theory. 2011. arXiv:1108.4876v1. https://arxiv.org/abs/1108.4876
- E. L. Mehler, The Lorentz-Debye-Sack theory and dielectric screening of electrostatic effects in proteins and nucleic acids, in Molecular Electrostatic Potentials: Concepts and Applications, Theoretical and Computational Chemistry Vol. 3, 1996
- Bauer, M. R.; Mackey, M. D. Electrostatic Complementarity as a Fast and Effective Tool to Optimize Binding and Selectivity of Protein–Ligand Complexes. J. Med. Chem. 2019, 62 (6), 3036–3050. https://doi.org/10.1021/acs.jmedchem.8b01925.
- Vinter, J. G. Extended Electron Distributions Applied to the Molecular Mechanics of Some Intermolecular Interactions. II. Organic Complexes. J. Comput. Aided. Mol. Des. 1996, 10 (5), 417–426. https://doi.org/10.1007/BF00124473.
- Cresset的核心技术. 墨灵格的博客. http://blog.molcalx.com.cn/2019/10/08/cresset-science-xed.html
- 肖高铿.借助重要的水加速基于结构的ATX抑制剂先导化合物优化.墨灵格的博客. http://blog.molcalx.com.cn/2021/08/24/accelerate-autotaxin-inhibitor-lead-optimization.html
7. 联系我们
想亲自试用Flare与SPARK,或技术合作请联系我们。


