
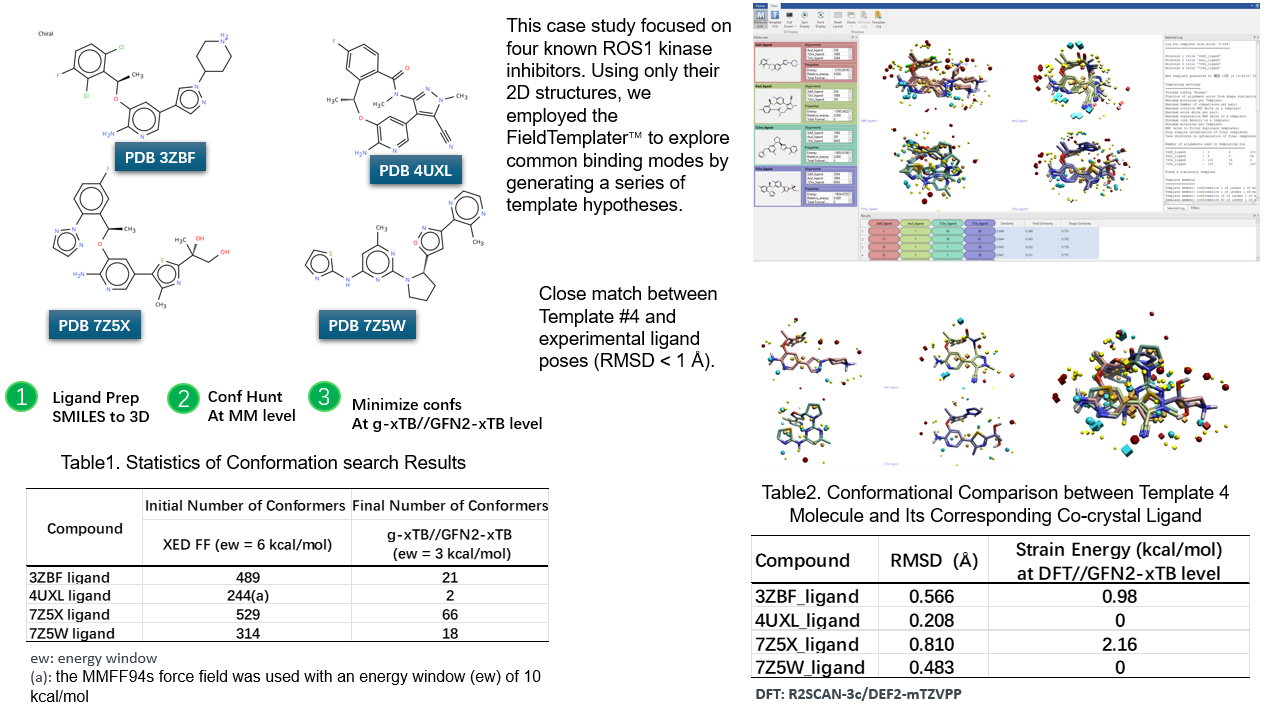
用FieldTemplater预测ROS1抑制剂的生物活性构象
摘要:在缺乏靶标蛋白共晶结构信息的早期药物发现阶段,准确识别配体的生物活性构象对虚拟筛选与先导化合物优化至关重要。本研究以四个已知ROS1激酶抑制剂为对象,在仅提供其二维化学结构的前提下,利用Cresset公司Flare™平台中的FieldTemplater™模块,基于分子静电场、疏水场与形状场的连续性特征,构建其公共结合模式。结果表明,FieldTemplater成功识别出高置信度模板(模板4),不仅在激酶铰链区展现出与ATP腺嘌呤片段高度一致的氢键及静电相互作用模式,且与实验共晶结构中的配体构象高度吻合(RMSD均低于0.81 Å)。构象张力能分析进一步证实,所预测构象在热力学上稳定, 在R2SCAN-c3/DEF2-mTZVPP//GFN2-xTB理论水平计算的张力能 ΔE \(<\) 2.16 kcal/mol 。本研究验证了FieldTemplater在无结构信息条件下可靠推断生物活性构象的能力,为基于配体的三维药效团建模与虚拟筛选提供了有效策略。
1. 前言
在前期研究1中,为确定化合物31(图1)的生物活性构象,我们利用Flare™平台中的场点(Field Points)技术,将其分别叠合至四个已知ROS1抑制剂与其靶标形成的共晶结构中的配体(图2)上。结果表明,化合物31的两种构象——CONF_34与CONF_41——不仅在三维形状与药效团特征上与上述共晶配体高度相似,且能够有效模拟这些配体与ROS1激酶铰链区及DFG基序关键残基之间的相互作用模式,因而被推测为其潜在的生物活性构象。
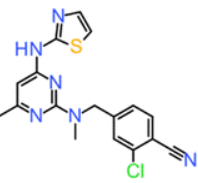
图1. 化合物31的化学结构式
此外,研究还识别出一种高张力能的“非活性”构象(CONF_17),其构象张力能高达8.5 kcal/mol,表明该构象在热力学上不稳定;同时,该构象缺乏与ROS1铰链区形成氢键相互作用所需的供体基团。随后,我们分别以活性构象(CONF_34/41)和非活性构象(CONF_17)作为查询分子,对ChEMBL35数据库开展基于形状的虚拟筛选。结果显示,以活性构象为模板的筛选不仅高效召回已知ROS1抑制剂,还能在最高排名中识别出潜在的反靶标警示信号2;相比之下,尽管非活性构象亦可召回部分已知抑制剂,但因其缺乏合理的生物相互作用特征,在后处理阶段被合理排除。上述结果进一步支持了所推测生物活性构象的合理性,并凸显了在虚拟筛选中采用正确生物活性构象作为查询分子的重要性。
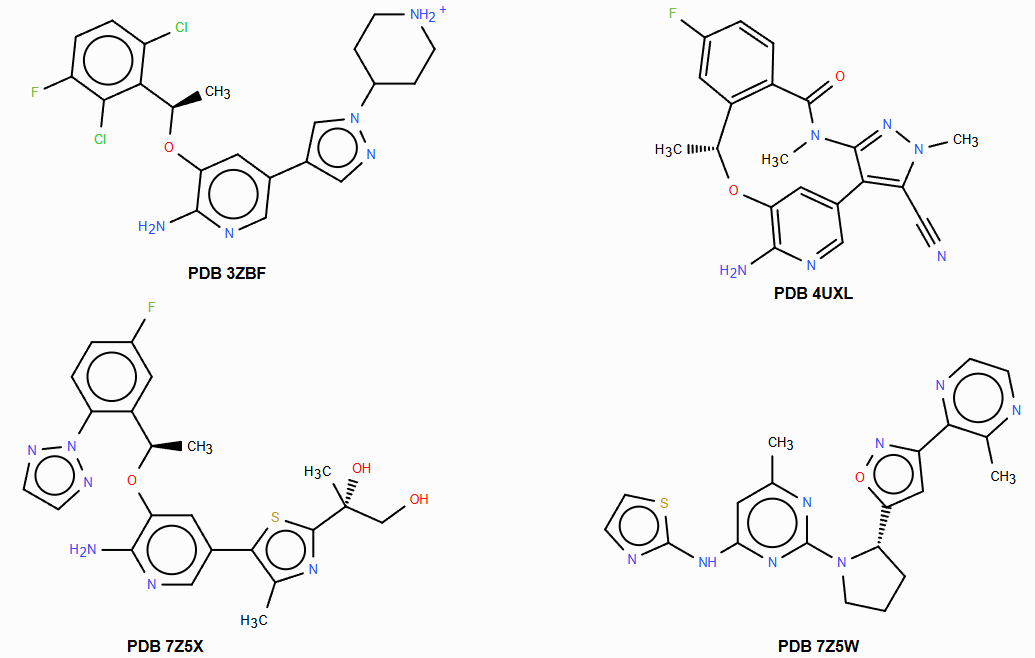
图2. 四个已知ROS1抑制剂及其与ROS1共晶结构的PDB代码
上述生物活性构象的推断依赖于四个ROS1抑制剂与靶蛋白的共晶结构信息。然而,在早期药物发现阶段,此类结构数据往往十分有限。在此情形下,基于配体的计算方法可作为基于结构方法的有效替代。Cresset公司开发的Flare™平台中的FieldTemplater™模块即为代表性工具之一3。FieldTemplater通过比较分子的静电场、疏水场及形状场,识别不同配体间潜在的公共结合模式。即使在缺乏靶标蛋白结构信息的前提下,该方法亦能从若干结构多样但作用于同一靶点的配体中,推导出其可能的生物活性构象及最优叠合方式。与传统药效团建模方法(通常依赖氢键供体/受体、疏水中心等离散特征)不同,FieldTemplater采用连续的分子场(包括静电、形状与疏水场)构建三维模板(Template)。此类模板可作为参考框架,用于指导先导化合物优化,或对虚拟化合物库中的分子进行系统性三维构象比对与筛选4。
本文旨在模拟早期药物发现场景——即仅基于图2中四个ROS1抑制剂的二维结构、而无共晶结构信息可用的情况下——利用FieldTemplater构建其公共结合模式,并将所得构象与实际共晶结构中的配体构象进行系统比对,从而对预测的生物活性构象进行评估与优化,并进一步验证FieldTemplater在无结构信息条件下识别活性构象的能力与可靠性。
2. 结果与讨论
2.1 构象搜索
本研究中,化合物的构象分析始于其SMILES二维结构表示。首先,采用Flare软件中的Ligand | Ligand Pred模块生成初始三维结构并进行初步几何优化。随后,利用3D Pose | Conf Hunt & Align模块构建构象系综。所有构象均在GFN2-xTB理论水平下进行几何优化,并执行g-xTB单点能计算。构象去重后,保留能量窗口为3 kcal/mol内的构象,具体方法详见3.1节“构象搜索”。与传统虚拟筛选流程相比,本研究在力场参数设置中特别启用了“Coulombic and attractive vdW”选项,以更准确地描述分子内氢键以及S···N/O超共轭相互作用的潜在贡献1。
值得注意的是,PDB 4UXL的共晶配体具有大环结构特征,因此采用自定义的Flare扩展应用Flare | Extensions | Macrocycle Conf Search进行处理:首先使用RDKit的ETKDGv3算法生成初始大环构象,随后采用MMFF94s力场进行几何优化,并以重原子RMSD = 0.5 Å作为构象重复性判据,最终保留能量窗口为10 kcal/mol内的构象系综。具体过程详见3.2节“大环分子的构象搜索”。
| Compound | 初始构象数 XED FF ew = 6 kcal/mol |
最终构象数 g-xTB//GFN2-xTB ew = 3 kcal/mol |
|---|---|---|
| 3zbf_ligand | 489 | 21 |
| 4uxl_ligand | 244a | 2 |
| 7z5x_ligand | 529 | 66 |
| 7z5w_ligand | 314 | 18 |
ew: energy window;a:4UXL配体采用MMFF94s力场,ew = 10 kcal/mol。
构象搜索的最终统计结果汇总于表1,所获得的构象系综将用于后续的活性构象预测研究。
2.2 用FieldTemplater生成模板:公共结合模式的预测
FieldTemplater的药效团识别基于以下假设:当多个配体以相似的结合模式作用于同一蛋白结合位点时,其分子场(包括静电、形状与疏水场)应呈现高度一致性。因此,FieldTemplater通过遍历输入配体的构象空间,识别其公共场模式。该公共场模式可解释结构多样化合物共享的结合机制。在分子叠合的初始阶段,系统基于场点评估分子间的共性,随后通过全场相似性计算进行优化。每个生成的药效团模型为每个输入分子指定一个活性构象,该模型即构成一个“模板”(Template)。在本研究中,“药效团模型”与“模板”为同义术语;需特别指出的是,FieldTemplater中的药效团元素为连续的场点(Field Points,图3),而非传统药效团中离散的氢键供体/受体、电荷中心或疏水区域等特征。
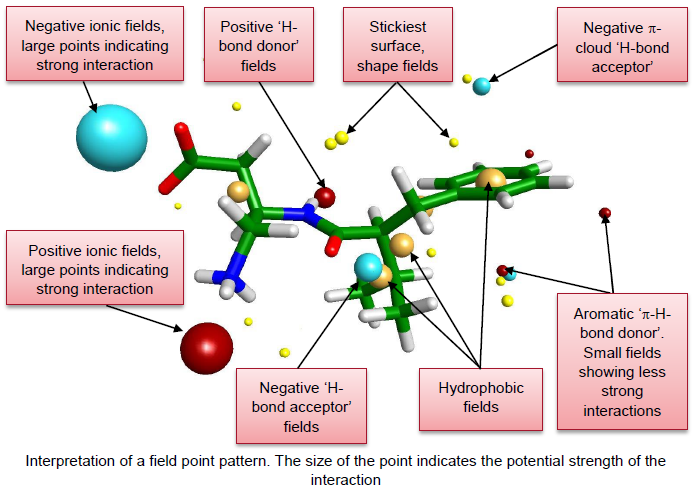
图3. 场点示意图
在Flare的FieldTemplater模块中,导入前述生成的四个化合物构象系综,在标准模式下执行计算,主要参数设置如下:
- 最大构象数:100;
- Similarity打分中形状相似性权重:50%;
- 每对化合物间最大比较次数:100;
- 每对化合物间最大Δ打分阈值:0.10。
共生成217个模板,按Similarity值排序后,前四个最优模板在结果表格中被高亮显示,并在3D视窗中可视化呈现,如图4所示。
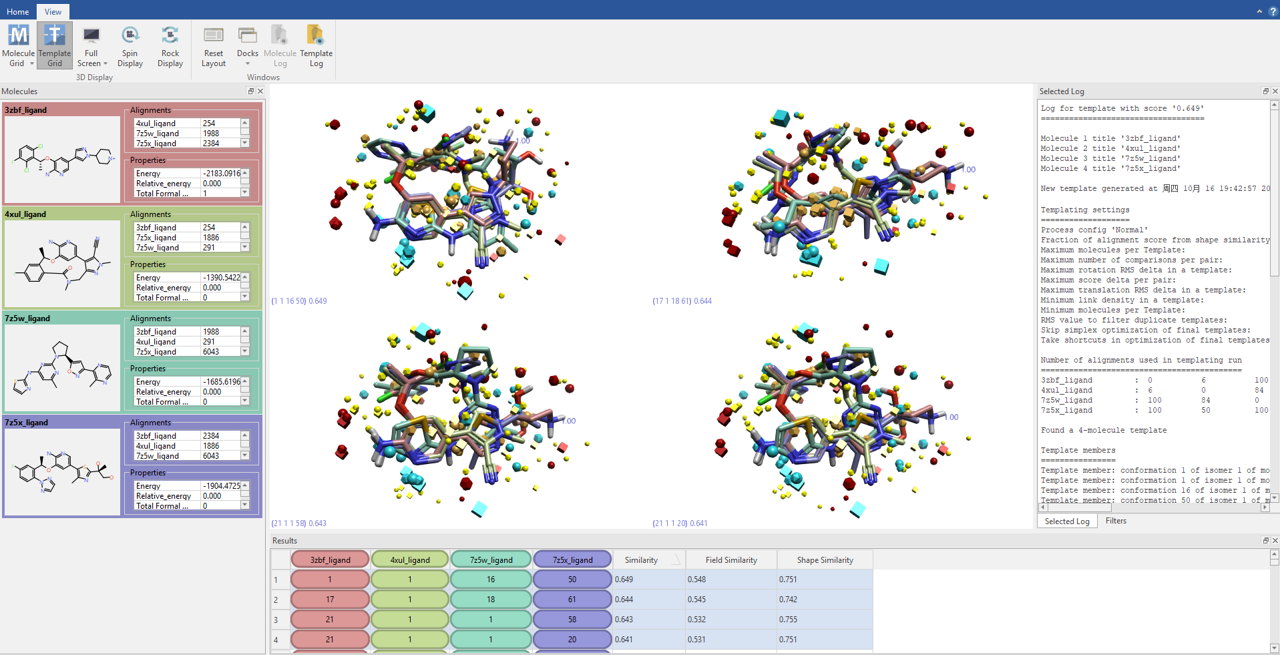
图4. 四个ROS1抑制剂经FieldTemplater计算所得的前四个模板(默认参数)
2.3 模板的选择与评估:FieldTemplater重现了ROS1抑制剂的生物活性构象
模板的选择基于两个关键标准:
高相似性打分(Similarity值):首先对所有217个模板按Similarity值降序排列,优选考虑Similarity值高的;
符合激酶铰链区的典型结合模式:具备与激酶铰链区的经典相互作用特征,优先选择能够同时覆盖全部四个ROS1抑制剂的模板。
首先,根据Similarity值对模板进行降序排列,并优先筛选能够同时涵盖全部四个分子的模板。排名靠前的120个模板均满足此条件。
其次,鉴于所有四个ROS1激酶抑制剂均结合于激酶的正构位点,我们假设其结合模式可模拟激酶底物ATP的腺嘌呤部分与铰链区gatekeeper残基形成经典氢键及C–H···O=C弱氢键相互作用6(图5):多数抑制剂通过其骨架上的氢键供体与受体,分别与gatekeeper+3(gk+3)和gatekeeper+1(gk+1)位置的羰基氧及酰胺氮形成氢键。此外,C–H···O=C相互作用可通过Flare的场点分析进一步识别7。
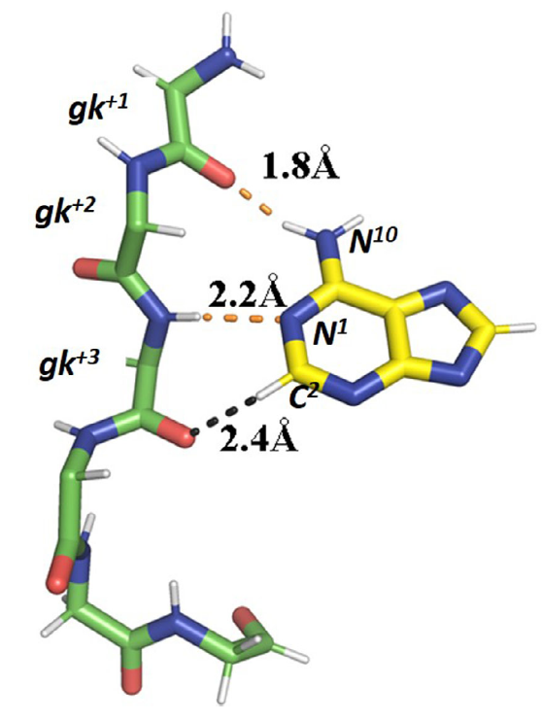
图5. 以AMPPNP与RSK2 N-端结构域的复合物晶体结构(PDB: 3G51,分辨率1.8 Å)为例,展示ATP腺嘌呤部分与激酶铰链区gatekeeper残基之间的氢键相互作用6
基于上述铰链区相互作用模式的先验知识,可快速识别出打分靠前的模板3、4、5具备生物学意义的候选。由于从视觉上看,模板3、4、5看着似乎相差不大,因此接下来以主要模板3、4为主来进一步考察。以如图6(上)所示,模板3与4中高亮的红色(氢键供体,HBD)–绿色(氢键受体,HBA)–红色(HBD)场点排列,与图5中ATP腺嘌呤的N6H2(HBD)–N1(HBA)–C2H(HBD)模式高度对应,且四个分子均满足该排列。
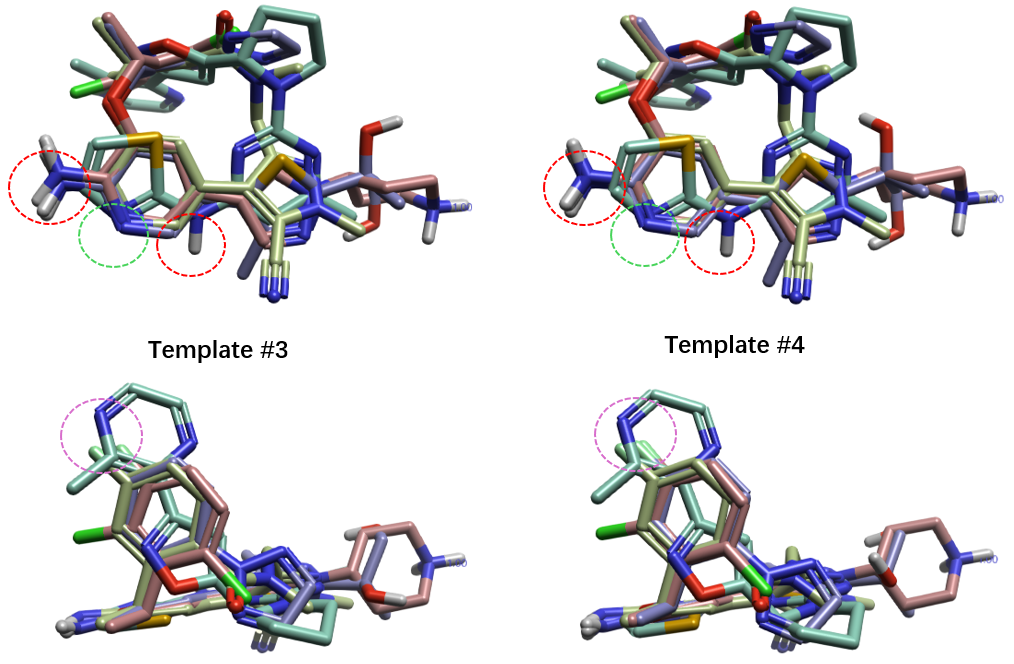
图6. 模板3(左)与模板4(右)的两个视角:上图为铰链区场点匹配,下图为DFG区芳香片段叠合
此外,如图6(下)所示,模板3与4还显示四个抑制剂指向DFG基序的芳香片段高度叠合,尤其是吡嗪环上的氮原子与苯环上的氟原子在空间上重合(紫色圆圈高亮区域),表明这些结构多样的分子在DFG方向上具有高度一致的静电特征。
相比之下,尽管模板1与模板2具有最高的总体Similarity值,但至少有一个分子无法满足铰链区氢键模式或DFG区静电一致性要求,因此被排除。

图7. FieldTemplater计算结果前6个模板的打分值及各个模板中每个分子的构象ID
图7给出了模板3、4、5的总体相似性(Similarity)、场相似性(Field Similarity)与形状相似性(Shape Similarity)的平均值以及每个模板中各个分子的构象ID。可以发现,尽管我们可以对各种相似性指标进行从高到低的排序,但无论从哪一个指标的数值上看,差异均极小: 仅在小数点后第三位有所区别。从构象ID上看,在模板3、4、5中,3ZBF、4UXL、7Z5W配体均采取相同的构象,仅7Z5X配体在三个模板中发生变化。
鉴于模板3、4、5在相似性指标上差异极小,有必要进一步考察组成模板的各配体构象能差异,并与共晶配体的构象进行比较,以评估其是否成功重现了实验结合模式。在Flare中,以每个模板中四个分子的预测构象为参考,分别与各自ROS1共晶结构中的配体进行刚性叠合(使用Flare的3D Pose | Superpose功能),计算二者之间的重原子RMSD,以评估预测构象与实验构象的一致性。通常认为RMSD \(<\) 2.0 Å可视为成功重现实验结合模式。结果如表2-4所示,模板3、4、5的四个配体的预测构象均高精度重现了其对应的生物活性构象(RMSD范围:0.208–1.160 Å)。其中,模板4的7Z5X配体不仅构象张力能(ΔE = 1.378 kcal/mol)最低,其RMSD值(0.810 Å)也为最低,因此被确定为最优模板。
| Compound | RMSD (Å) | Strain Energy (kcal/mol) at g-xTB//GFN2-xTB level |
ConfId |
|---|---|---|---|
| 3zbf_ligand | 0.566 | 2.965 | 21 |
| 4uxl_ligand | 0.208 | 0.0 | 1 |
| 7z5x_ligand | 1.160 | 2.758 | 58 |
| 7z5w_ligand | 0.483 | 0.0 | 1 |
| Compound | RMSD (Å) | Strain Energy (kcal/mol) at g-xTB//GFN2-xTB level |
ConfId |
|---|---|---|---|
| 3zbf_ligand | 0.566 | 2.965 | 21 |
| 4uxl_ligand | 0.208 | 0.0 | 1 |
| 7z5x_ligand | 0.810 | 1.378 | 20 |
| 7z5w_ligand | 0.483 | 0.0 | 1 |
| Compound | RMSD (Å) | Strain Energy (kcal/mol) at g-xTB//GFN2-xTB level |
ConfId |
|---|---|---|---|
| 3zbf_ligand | 0.566 | 2.965 | 21 |
| 4uxl_ligand | 0.208 | 0.0 | 1 |
| 7z5x_ligand | 1.024 | 2.880 | 61 |
| 7z5w_ligand | 0.483 | 0.0 | 1 |
2.4 在DFT//GFN2-xTB理论水平下评估构象的生物合理性
根据 Rai 等人9对 Cambridge Structural Database (CSD) 晶体结构及 Protein Data Bank (PDB) 共晶配体生物活性构象的系统性研究,在密度泛函理论(DFT)水平下,生物相关构象的全局构象张力能通常不超过 2.5 kcal/mol。该阈值可作为判断配体是否处于热力学合理活性构象的经验标准。基于此,尽管模板3、4、5在几何叠合上表现良好,但其预测的 3ZBF 与 7Z5X 配体构象在 g-xTB 水平下的张力能分别为 2.965 kcal/mol 与 2.880 kcal/mol,均已超出上述阈值,提示其作为生物活性构象的合理性存疑。值得注意的是,g-xTB 作为一种半经验方法,虽具备较高计算效率,但其精度介于 GFN2-xTB 与 DFT 之间,可能难以准确捕捉复杂分子体系中的细微能量差异。为更可靠地评估构象稳定性,我们采用更高精度的 R2SCAN-3c/mTZVPP 理论方法重新计算构象张力能。该方法在保持合理计算成本的同时,兼具高精度与速度,已被推荐为几何优化与能量计算的首选方案之一10。
在此基础上,我们再次考察了三个结构高度相似的候选模板——模板3、4 与 5。这三个模板的差异仅体现在 7Z5X 配体的构象上,其余三个配体(3ZBF、4UXL、7Z5W)在所有模板中均保持完全一致的构象。因此,该比较可有效隔离 7Z5X 构象变化对整体模型质量的影响,从而聚焦于其构象合理性评估。计算结果汇总于表5–7。
结果显示,4UXL 与 7Z5W 配体的预测构象张力能均为 0.00 kcal/mol,表明其处于能量极小点;3ZBF 配体的张力能由 g-xTB 计算的 2.965 kcal/mol 显著降低至 0.98 kcal/mol,远低于 2.5 kcal/mol 的生物相关性阈值,确认其预测构象在热力学上高度可行。相比之下,7Z5X 配体的构象张力能在不同模板中表现出显著差异:在模板3 与模板5 中,其值分别高达 3.90 kcal/mol 与 3.89 kcal/mol,均明显超出合理范围;而在模板4 中,该值降至 2.16 kcal/mol,低于 2.5 kcal/mol 阈值,符合生物活性构象的能量特征。这一结果表明,模板4 所采用的 7Z5X 构象在热力学上更为合理,是更可信的活性构象候选。
如上一小节所述,模板预测的结合构象与实验共晶结构进行刚性叠合的重原子 RMSD 分析显示,模板4 中 7Z5X 配体的 RMSD 为 0.810 Å,显著低于模板3(1.160 Å)与模板5(1.024 Å),表明其空间取向最近实验观测的生物活性构象。需要强调的是,RMSD 在此仅作为回溯性验证指标,而非模型选择依据;真正的判别标准在于构象张力能是否满足生物相关性阈值。综合来看,模板4 不仅在能量上符合药物化学对低张力活性构象的要求,其预测结果亦得到实验结构的有力支持。因此,在所有候选模型中,模板4 展现出最优的综合性能,可作为可靠的三维药效团参考模型,用于后续的虚拟筛选、骨架跃迁及先导化合物优化。
| Compound | RMSD (Å) | Strain Energy (kcal/mol) at DFT//GFN2-xTB level |
ConfId |
|---|---|---|---|
| 3zbf_ligand | 0.566 | 0.98 | 21 |
| 4uxl_ligand | 0.208 | 0.00 | 1 |
| 7z5x_ligand | 1.160 | 3.90 | 58 |
| 7z5w_ligand | 0.483 | 0.00 | 1 |
DFT: R2SCAN-3c/mTZVPP
| Compound | RMSD (Å) | Strain Energy (kcal/mol) at DFT//GFN2-xTB level |
ConfId |
|---|---|---|---|
| 3zbf_ligand | 0.566 | 0.98 | 21 |
| 4uxl_ligand | 0.208 | 0.00 | 1 |
| 7z5x_ligand | 0.810 | 2.16 | 20 |
| 7z5w_ligand | 0.483 | 0.00 | 1 |
DFT: R2SCAN-3c/mTZVPP
| Compound | RMSD (Å) | Strain Energy (kcal/mol) at DFT//GFN2-xTB level |
ConfId |
|---|---|---|---|
| 3zbf_ligand | 0.566 | 0.98 | 21 |
| 4uxl_ligand | 0.208 | 0.00 | 1 |
| 7z5x_ligand | 1.024 | 3.89 | 61 |
| 7z5w_ligand | 0.483 | 0.00 | 1 |
DFT: R2SCAN-3c/mTZVPP
3. 方法
3.1 构象搜索
分子的初始三维构象由其SMILES表示出发,首先利用Flare软件中的Ligand | Ligand Pred模块生成初始3D结构,并进行初步几何优化。随后,采用3D Pose | Conf Hunt & Align模块生成构象系综,具体参数设置如下:
- Conformation hunt: Very Accurate and Slow
- Maximum number of conformations: 1000
- No. of high-T dynamics runs for flexible ring: 20
- Energy window: 6 kcal/mol
- Turn off Coulombic and attractive vdW forces: No
- Remove boats and twist-boats: No
所得构象系综进一步在Flare平台下通过3D Pose | QM | Minimize Conformation模块,在GFN2-xTB理论水平(气相,无溶剂模型)下进行几何优化。
经GFN2-xTB优化后的构象,采用g-xTB8方法进行单点能计算。g-xTB 是 Grimme 课题组最近推出的 GFN2-xTB 的替代方法8,其中”g”代表”通用”。该方法展现出显著优于GFN2-xTB的计算结果前景。基于g-xTB单点能结果,对构象进行聚类分析:若任意两个构象之间的相对能量差小于0.05 kcal/mol,且重原子RMSD小于0.125 Å,则视为重复构象,仅保留其中能量较低者。
3.2 大环分子的构象搜索
大环分子的构象搜索采用自定义的Flare扩展应用5(Flare | Extensions | Macrocycle Conf Search),参数设置如图8所示。
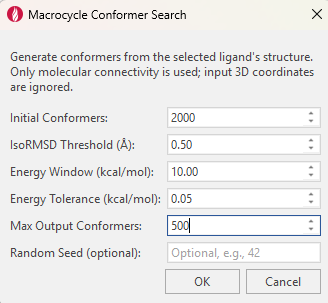
图8. 大环分子构象搜索参数
该拓展应用从分子的2D结构(SMILES表示)出发,使用RDKit的ETKDGv3算法生成初始大环构象,随后采用MMFF94s力场进行几何优化,并以重原子RMSD = 0.5 Å作为构象重复性判据,最终保留能量窗口为10 kcal/mol内的构象系综。生成的构象系综进一步通过3D Pose | QM | Minimize Conformation模块,在GFN2-xTB理论水平(气相,无溶剂模型)下进行几何优化,并采用g-xTB方法进行单点能计算。构象去重标准同3.1节。
4. 结论
本研究在无靶标蛋白结构信息的条件下,仅基于四个ROS1抑制剂的二维化学结构,成功利用FieldTemplater构建了其公共结合模式。所识别的模板4不仅在激酶铰链区准确再现了ATP腺嘌呤片段的经典氢键与非经C–H···O=C弱氢键相互作用,还在DFG区域展现出一致的静电特征。更重要的是,预测构象与实验共晶结构中的配体构象高度一致(RMSD \(≤\) 0.81 Å),且构象张力能较低,在R2SCAN-c3/DEF2-mTZVPP//GFN2-xTB理论水平计算的张力能 ΔE \(<\) 2.16 kcal/mol ,表明其具备热力学可行性。该结果充分验证了FieldTemplater在早期药物发现中推断生物活性构象的可靠性与实用性。因此,基于XED力场的FieldTemplater方法可作为传统离散药效团模型的有效补充,为无靶标结构信息场景下的虚拟筛选、先导化合物优化及作用机制解析提供强有力的计算支持。
5. 接下来可以做什么?
- 将模板分子作为3D-QSAR的参比分子,用于指导3D分子叠合
- 将模板分子用于基于形状虚拟筛选,比如ROCS,FastROCS
- 将模板分子用于基于XED场的虚拟筛选,比如Flare align与Blaze虚拟筛选
6. 附件
文件链接: ROS1-inhibitor-fieldtemplater 提取码: q67g
- FieldTemplater.flr: Flare项目文件,包含了配体的构象系综、共晶配体以及FieldTemplater结果等
- 3zbf_relative_energy.csv:3zbf配体构象系综的相对能量
- 4uxl_relative_energy.csv:4uxl配体构象系综的相对能量
- 7z5x_relative_energy.csv:7z5xl配体构象系综的相对能量
- 7z5w_relative_energy.csv:7z5w配体构象系综的相对能量
7. 文献
- 查寻分子的准备对基于形状虚拟筛选的影响. http://blog.molcalx.com.cn/2025/10/11/the-impact-of-query-conformation-on-shape-based-virtual-screening.html
- 化合物31的化学基因组学分析——基于3D形状相似性的靶标预测研究. http://blog.molcalx.com.cn/2025/10/14/shape-based-target-fishing-for-compound-31.html
- FieldTemplater. https://cresset-group.com/software/fieldtemplater
- Timothy S. Chisholm, Mark Mackey, and Christopher A. Hunter. Discovery of High-Affinity Amyloid Ligands Using a Ligand-Based Virtual Screening Pipeline. Journal of the American Chemical Society. 2023,145(29): 15936-15950. DOI: 10.1021/jacs.3c0374
- macrocycle_confgen.py. https://github.com/gkxiao/pyflare-extension
- Derewenda, Z. S.; Hawro, I.; Derewenda, U. C─H⋯O Hydrogen Bonds in Kinase-Inhibitor Interfaces. IUBMB Life 2020, 72 (6), 1233–1242. https://doi.org/10.1002/iub.2282.
- 用XED力场分析激酶抑制剂的C─H···O=C氢键. 墨灵格的博客. http://blog.molcalx.com.cn/2022/01/31/c-h-o-hydrogen-bonds-in-kinase-inhibitor.html
- Froitzheim, T. et al. (2025) “g-xTB: A General-Purpose Extended Tight-Binding Electronic Structure Method For the Elements H to Lr (Z=1–103).” Available at: https://doi.org/10.26434/chemrxiv-2025-bjxvt.
- Bursch, M. et al. (2022) “Best‐Practice DFT Protocols for Basic Molecular Computational Chemistry,” Angewandte Chemie International Edition, 61(42). Available at: https://doi.org/10.1002/anie.202205735.
- Rai, B.K. et al. (2019) “Comprehensive Assessment of Torsional Strain in Crystal Structures of Small Molecules and Protein–Ligand Complexes using ab Initio Calculations,” Journal of Chemical Information and Modeling, 59(10), pp. 4195–4208. Available at: https://doi.org/10.1021/acs.jcim.9b00373.


