
用XED力场分析激酶抑制剂的C─H···O=C氢键
摘要:量子化学计算表明,C─H···O氢键相互作用强度相当强,大约是N─H···O氢键相互作用强度的一半。最近的研究表明,几乎所有的FDA批准上市的激酶抑制剂均与激酶铰链存在C─H···O型氢键相互作用。经典的氢键相互作用不是激酶抑制剂与铰链相互作用所必须的,也可仅通过C─H···O型氢键相互作用而保留抑制剂的活性。Flare的XED静电分析工具可以帮我们非常方便地分析这种非经典的C─H···O氢键相互作用。
肖高铿/2022/01/25
1. XED力场概述
静电势在配体 – 受体识别与结合过程中发挥关键作用,然而因其计算复杂度较高且可视化方法相对繁琐,该因素在早期分子设计研究中常被低估。既往研究表明,静电信息可高效应用于骨架跃迁(scaffold hopping)、虚拟筛选、配体构象叠合及构效关系(SAR)解析等关键环节[1]。具体而言,配体的静电特征可通过引入探针原子,系统计算其与配体构象在三维空间中的相互作用能而获得;该相互作用能定量表征了空间任意位点的静电属性。如图 1 所示,静电势可进一步以等值面形式进行可视化呈现。Cresset 公司开发的 XED(Extended Electron Distribution)力场采用原子偏心电荷模型,能够精确刻画分子的静电分布特征[1]。该力场的核心优势在于可将部分电荷的π与σ组分进行解耦处理,从而更真实地反映分子轨道的电子分布特性。
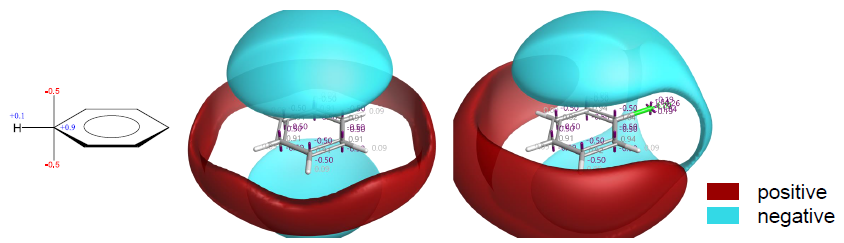
图1. 苯分子的 XED 模型、外部点电荷分布及其静电可视化
除配体静电性质的计算外,XED 力场亦适用于受体蛋白静电场的表征,以及配体 – 受体间静电互补性分析与蛋白 – 配体相互作用能评估等分子设计关键任务[2,3]。本文旨在应用该力场,系统解析激酶抑制剂中 C─H···O=C 型非经典氢键的相互作用。
2. C─H···O=C氢键相互作用
蛋白激酶作为重要的药物靶点,其抑制剂可广泛应用于神经退行性疾病、炎症反应、心血管疾病及恶性肿瘤等重大疾病的治疗。据 Derewenda 等[4]统计,截至 2020 年,美国食品药品监督管理局(FDA)已批准约 48 种激酶抑制剂上市,其中抗肿瘤适应症占比最高,其余主要集中于炎症与自身免疫性疾病领域。激酶抑制剂研发的核心挑战在于选择性优化:人类激酶组包含 517 种蛋白激酶,其催化结构域具有高度序列与结构同源性,尤其是多数抑制剂靶向的 ATP 结合口袋,表现出显著的氨基酸组成与三维构象保守性。
目前上市的激酶抑制剂多为 I 型抑制剂,即通过竞争性占据 ATP 结合位点发挥药理作用。激酶口袋对腺嘌呤的特异性识别依赖于一个特征性的氢键网络,该网络涉及一段暴露于溶剂、被称为”铰链区(hinge region)”的短肽片段,其连接激酶催化结构域的 N 端与 C 端亚结构域。铰链区通常由三个氨基酸残基构成,呈延伸的β-折叠二级结构构象;依据其相对于”看门人(gatekeeper)”残基的序列位置,依次标记为 gk+1、gk+2与 gk+3。ATP 分子中腺嘌呤环与铰链区的结合模式为:腺嘌呤 N1位氮原子作为氢键受体,与 gk+1残基的主链酰胺氮形成氢键;N6位氨基作为氢键供体,与 gk+3残基的主链羰基氧形成氢键;此外,还存在一个非经典氢键相互作用,即腺嘌呤 C2-H 基团作为弱供体,与 gk+3残基的主链羰基氧形成 C─H···O 型氢键(图 2)[4]。
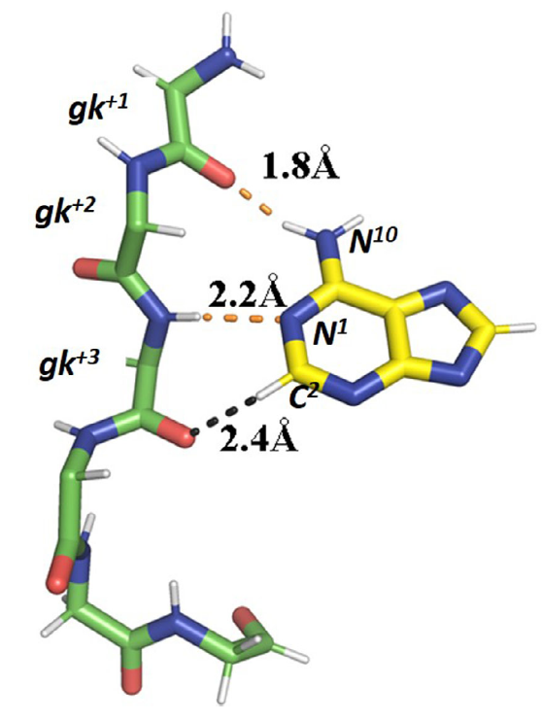
图2. 以 AMPPNP 与 RSK2 N 端结构域复合物晶体结构(PDB: 3G51,分辨率 1.8 Å)为例,展示 ATP 腺嘌呤片段与激酶铰链区肽段间的氢键相互作用网络[4]<
多数激酶抑制剂通过模拟腺嘌呤 – 铰链区的结合模式,利用氢键供体/受体基团与 gk+1、gk+3位点的羰基及酰胺基团形成经典氢键相互作用。尽管 Xing 等[7]已对上述典型氢键模式进行了系统分析,但其研究范围主要限于涉及电负性原子(如 N、O)作为供体/受体的经典相互作用。值得注意的是,多项文献报道在激酶 – 抑制剂复合物中存在 C─H···O=C 型非经典氢键(亦称”弱氢键”)[7,8],然而针对该相互作用的系统性研究仍较为有限。Derewenda 等[4]对 29 种已上市激酶抑制剂与其靶标蛋白的共晶结构进行了系统分析,发现抑制剂分子与铰链区羰基氧之间普遍存在 C─H···O=C 氢键相互作用。此外,作者指出,当前获批激酶抑制剂的骨架类型相对有限,且在利用铰链区羰基作为氢键受体方面呈现高度一致性:该羰基不仅可作为经典氢键的受体,亦可作为弱氢键的受体,其中 C─H 供体通常来源于芳环体系或与电负性氮原子相邻的亚甲基/次甲基基团。
研究初期,作者从蛋白质数据库(PDB)中筛选出 31 个相关晶体结构,其中包括 21 个 I 型抑制剂(ATP 竞争性)、7 个 II 型抑制剂(结合非活性构象)及 3 个 VI 型抑制剂(共价不可逆型)。除共价抑制剂 cobimetinib 因未与铰链区形成直接接触而被排除外,其余 30 个抑制剂 – 激酶复合物结构均纳入后续分析(图 3)[4]。
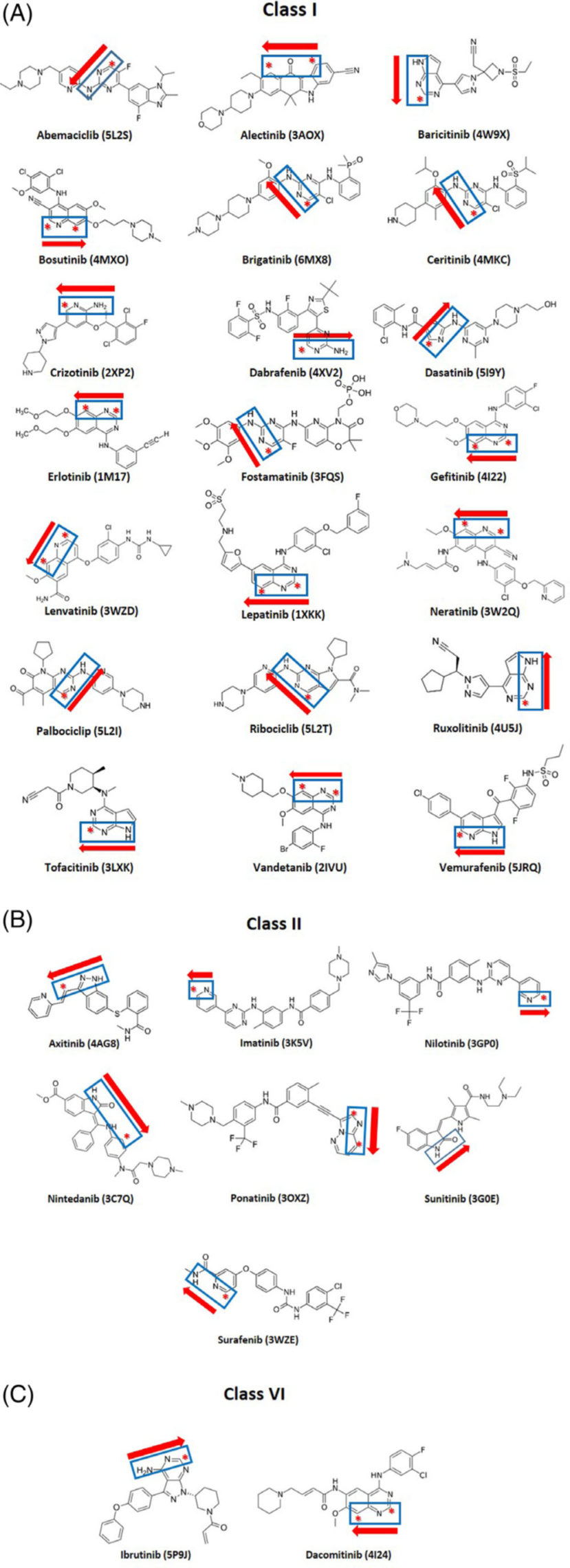
图 3. 30 种已上市激酶抑制剂的化学结构式。蓝色框标示与激酶铰链区形成氢键相互作用的供体/受体原子;红色星号标注作为 C─H···O=C 氢键供体的碳原子;箭头指示氢键基团相对于铰链区的空间取向(方向由 gk+1指向 gk+3)。(A) I 型抑制剂;(B) II 型抑制剂;(C) VI 型抑制剂。图片来源:文献 [4]。
综上所述,C─H···O=C 非经典氢键在已上市激酶抑制剂中具有普遍性。本文的核心目标在于借助 Flare 平台集成的 XED 力场,对该类弱氢键相互作用进行定量分析与可视化表征,以期为激酶抑制剂的理性设计提供理论依据与方法学支持。
3. 用XED力场发现、可视化分析C─H···O=C氢键相互作用
3.1 可视化分析CDK6抑制剂的C─H···O=C氢键
Flare提供了配体表面计算工具(见图4)可以用分析正、负静电势、疏水势以及势差。氢键的本质是静电相互作用,因此可借助Flare的XED力场来进行可视化分析。
图4. Flare提供了配体的表面分析工具,包括正、负XED力场的静电分析工具。
以上市的CDK6激酶抑制剂Ribociclib为例,其共晶结构为PDB 5L2T。在Flare里下载共晶结构,用protein prep进行标准的结构准备,接着用Contact分析相互作用,再用LIGAND选项卡下的+ev分析配体的正静电势,如图5所示。
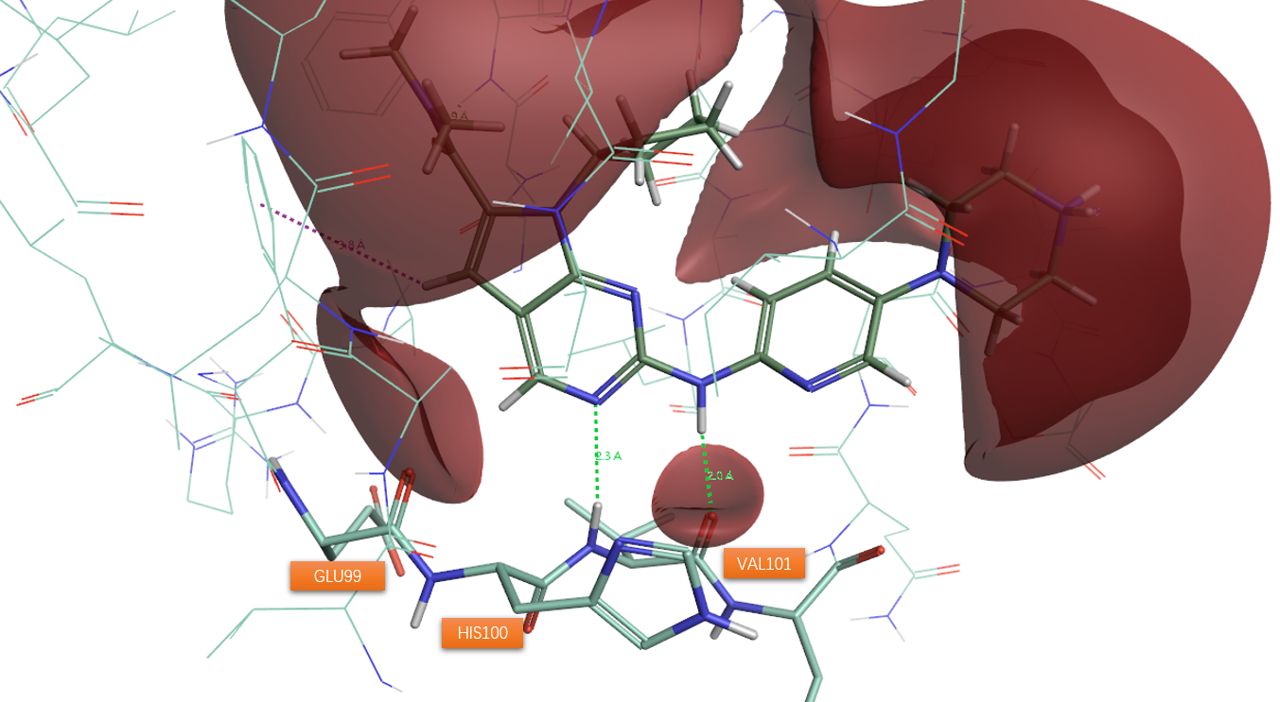
图5. Ribociclib与CDK6的相互作用示意图。其中红色等值图XED静电相互作用2.0等值面。
如图5所示,抑制剂Ribociclib与CDK6铰链gk+3残基VA101的酰胺NH供体以及羰基氧受体发生了经典的氢键相互作用。在沿着配体连接两环的N-H氢键供体的方向上,有着一个红色区块与VAL101羰基氧重合,这与配体N-H···O=C-Val氢键相互作用是一致的。配体嘧啶环上的氮作为氢键受体与HIS100的酰胺NH发生经典的氢键相互作用。在配体嘧啶环受体N邻位C-H方向上,有一个红色正静电势区块与蛋白的gk+1残基GLU99羰基氧重合,这意味着这这个原子对上发生了C-H···O=C非经典氢键相互作用。
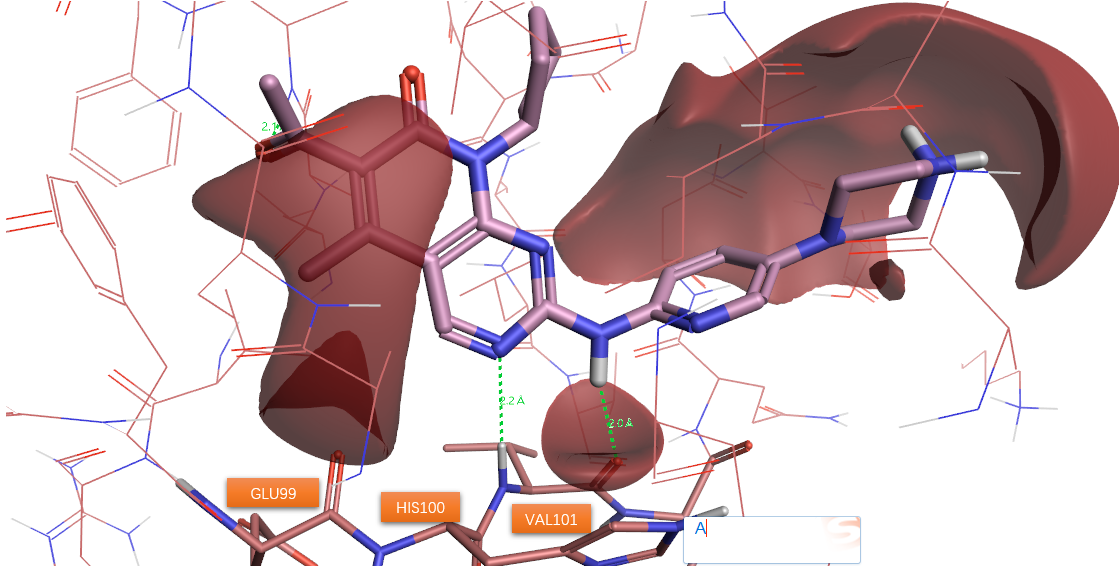
图6. Palbociclib与CDK6的相互作用示意图。其中红色等值图XED静电相互作用2.0等值面。
在另一个CDK6抑制剂Palbociclib上,我们也观察到了相似的作用模式。如图6所示,分析共晶结构5L2I,铰链区gk+1残基VAL99的羰基氧与配体的正静电势重合,而这正静电势正是在配体的嘧啶环N邻位C-H轴向上,因此可认为发生了C-H···O=C氢键相互作用。配体的嘧啶环N作为受体与gk+2残基HIS100的酰胺NH发生经典的氢键相互作用。而配体连接两环的NH作为供体,与铰链gk+3残基VAL101的羰基氧发生经典的N-H···O=C氢键相互作用。
3.2 可视化分析CDK2抑制剂的C─H···O氢键
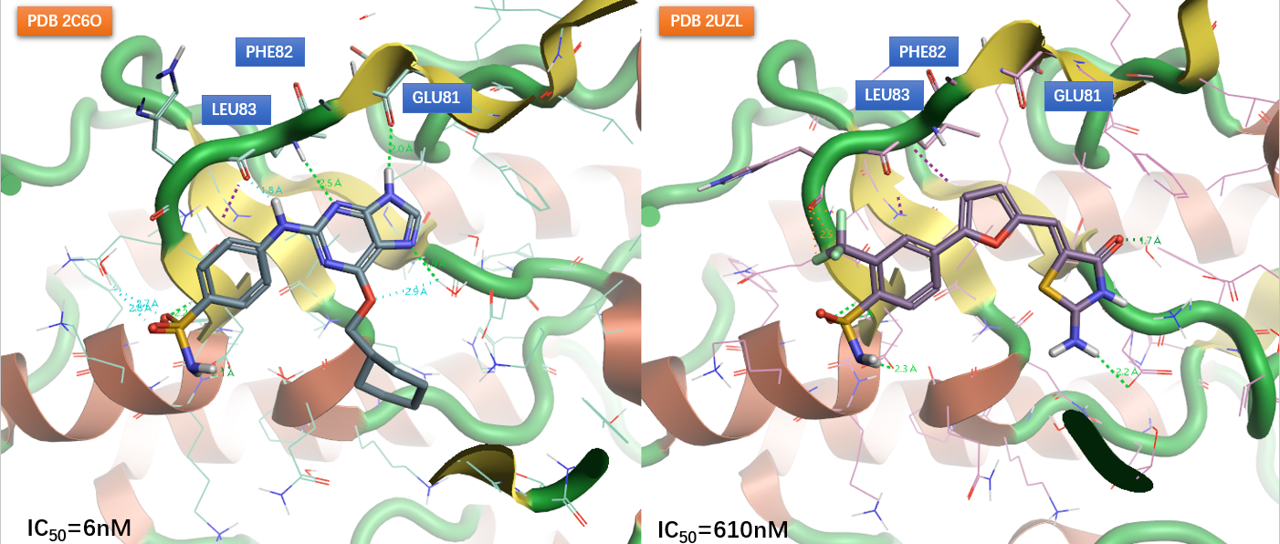
图7. CDK2抑制剂相互作用模式
图7左展示了一个CDK2抑制剂与靶标之间复合结构的相互作用模式(PDB 2C6o)。该抑制剂的一个连接两环的NH作为氢键供体与铰链gk+3残基LEU83的羰基氧发生氢键相互作用;一个芳香氮作为受体与gk+2残基LEU82的酰胺NH发生氢键相互作用;还有一个配体咪唑环上的NH作为供体与gk+1残基GLU81的羰基氧发生氢键相互作用。相比之下,以经典氢键相互作用模式观察PDB 2UZL的配体,看起来该抑制剂与激酶铰链之间不存在任何的氢键相互作用,如图7右所示。
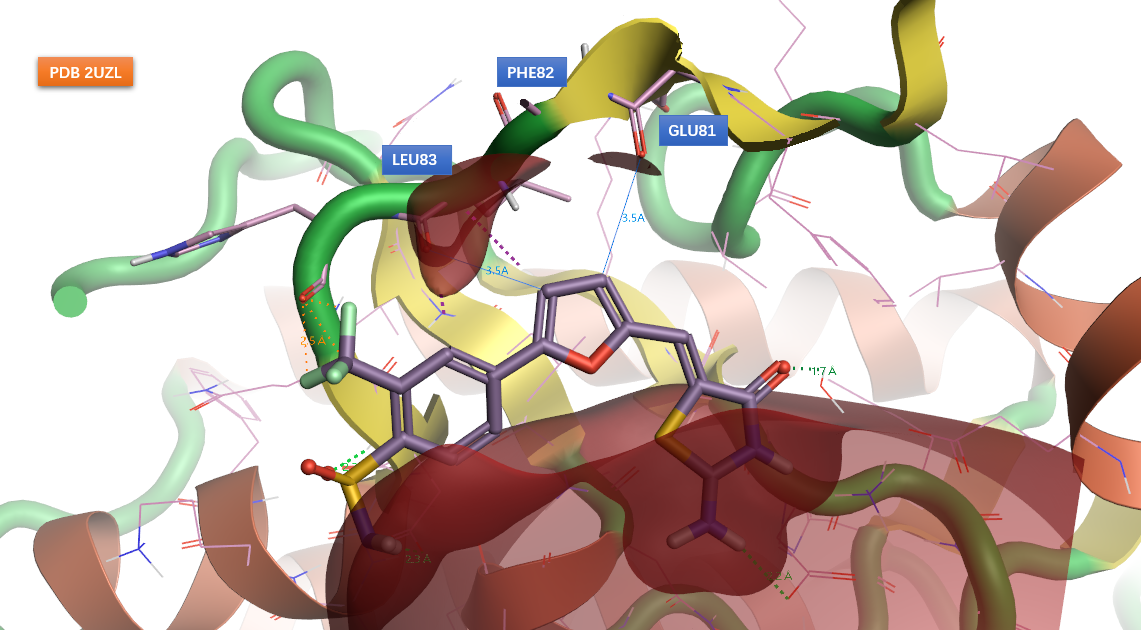
图8. PDB 2UZL的CDK2抑制剂静电
但是借助XED静电场,如图8所示,可以看到在沿着呋喃环的两个C-H轴向上,配体的两个正静电势区分别与铰链的gk+1残基GLU81、gk+3残基LEU83的主链羰基氧重合;并且从碳到氧的距离均为3.5Å,处于发生接触的距离之内。因此我认为配体呋喃环C-H与gk+1、gk+3的羰基氧发生了C─H···O=C氢键相互作用。根据Vargas等人[11]的量子化学计算研究,在真空中C─H···O=C氢键相互作用强度大约是N-H···O氢键相互作用强度的一半;此外,2UZL的配体比2C6O的配体少了一个与gk+1酰胺NH之间的氢键相互作用;这两个因素可能是前者比后者活性差的主要原因。2UZL的算例也说明了,I型抑制剂与铰链之间经典的氢键相互作用模式不是必须的。
4. 小结
量子化学计算表明,C─H···O=C氢键相互作用强度相当强,大约是N─H···O氢键相互作用强度的一半。几乎所有的FDA批准上市激酶抑制剂均与激酶铰链存在C─H···O=C型氢键相互作用。经典的氢键相互作用不是激酶抑制剂与铰链相互作用所必须的,也可仅通过C─H···O=C型氢键相互作用而保留抑制剂的活性。Flare的XED静电分析工具可以帮我们非常方便地分析这种非经典的C─H···O=C氢键相互作用。
5. 拓展应用
化合物的ADME与分子的lipinski氢键受体与供体数相关,经典的氢键受体/供体数量越大,极性表面积也越大,吸收、血脑屏障透过性越差。通过C─H···O=C氢键相互作用代替经典的氢键相互作用可以实现保留活性而改善化合物的ADME,尤其适合于先导化合物优化。
Flare提供了python界面,可以批量的计算一个氢键受体或供体处配体的静电势以便快速、高通量地进行分子对接后处理、将满足C─H···O=C氢键相互作用配体的过滤与识别出来[12]。
还可以借助BLAZE或SPARK软件,将XED静电用于高通量虚拟筛选,发现新型骨架先导化合物。
6. 文献
- Vinter, J. G. Extended Electron Distributions Applied to the Molecular Mechanics of Some Intermolecular Interactions. J. Comput. Aided. Mol. Des. 1994, 8 (6), 653–668. https://doi.org/10.1007/BF00124013.
- Cheeseright, T.; Mackey, M.; Rose, S.; Vinter, A. Molecular Field Extrema as Descriptors of Biological Activity: Definition and Validation. J. Chem. Inf. Model. 2006, 46 (2), 665–676. https://doi.org/10.1021/ci050357s.
- Bauer, M. R.; Mackey, M. D. Electrostatic Complementarity as a Fast and Effective Tool to Optimize Binding and Selectivity of Protein–Ligand Complexes. J. Med. Chem. 2019, 62 (6), 3036–3050. https://doi.org/10.1021/acs.jmedchem.8b01925.
- Derewenda, Z. S.; Hawro, I.; Derewenda, U. C─H⋯O Hydrogen Bonds in Kinase-Inhibitor Interfaces. IUBMB Life 2020, 72 (6), 1233–1242. https://doi.org/10.1002/iub.2282.
- Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S.The protein kinase complement of the human genome. Science. 2002;298:1912–1934.
- Fabbro D, Cowan-Jacob SW, Mobitz H, Martiny-Baron G.Targeting cancer with small-molecular-weight kinase inhibitors. Methods Mol Biol. 2012;795:1–34.
- Xing L, Klug-Mcleod J, Rai B, Lunney EA. Kinase hinge binding scaffolds and their hydrogen bond patterns. Bioorg Med Chem. 2015;23:6520–6527
- Pierce AC, Sandretto KL, Bemis GW. Kinase inhibitors and the case for CH…O hydrogen bonds in protein-ligand binding. Proteins. 2002;49:567–576.
- Pierce, A. C.; Ter Haar, E.; Binch, H. M.; Kay, D. P.; Patel, S. R.; Li, P. CH⋯O and CH⋯N Hydrogen Bonds in Ligand Design: A Novel Quinazolin-4-Ylthiazol-2-Ylamine Protein Kinase Inhibitor. J. Med. Chem. 2005, 48 (4), 1278–1281. https://doi.org/10.1021/jm0492249.
- Panigrahi SK. Strong and weak hydrogen bonds in protein ligand complexes of kinases: a comparative study. Amino Acids. 2008;34:617–633.
- Vargas, R.; Garza, J.; Dixon, D. A.; Hay, B. P. How Strong Is the C α −H···OC Hydrogen Bond? J. Am. Chem. Soc. 2000, 122 (19), 4750–4755. https://doi.org/10.1021/ja993600a.
- 肖高铿. 分子对接后处理——与指定残基发生特定相互作用配体的过滤. 墨灵格的博客. 2021-11-02. http://blog.molcalx.com.cn/2021/11/02/docking-post-filtering.html


