
摘要:Wang等人报道传统构象生成方法(如ConfGenX、OMEGA、RDKit)在A1O分子生物活性构象预测中完全失效,归因于方法学局限性,并建议优先采用人工智能模型。本文通过参数机制分析发现:失效主因是默认力场刻意忽略分子内长程相互作用项。具体而言,OMEGA默认力场mmff94smod_noestat排除库仑作用,Flare/XedeX默认关闭"长程静电与吸引性范德华力"选项,导致π-π堆积等关键作用未被描述。实验验证表明:当Flare启用完整非键相互作用项后,成功生成RMSD=0.96 Å的构象(精度超越文献报道的AI模型);OMEGA采用含完整静电项的mmff94smod力场亦可复现生物活性构象。本研究证实,传统方法在合理配置长程相互作用参数时,仍可高效预测强分子内作用体系的生物活性构象。该发现强调:构象生成方法的基准评测必须审慎验证力场参数设置,避免因参数配置偏差低估传统算法潜力。
肖高铿/2025-11-20
Wang等人1在评估传统方法与人工智能方法的小分子构象生成性能时发现:在数据集I(含3,354个高质量配体生物活性构象)的测试中,传统方法(包括ConfGenX、OMEGA、Conformator和RDKit ETKDG)对128个分子完全无法复现其生物活性构象。
A1O分子(图1,原文图4-B)是其中最具代表性的案例。A1O分子的晶体构象显示,其两个苯环通过π-π相互作用精确定位,这种构象在蛋白结合状态下稳定存在。然而,传统方法生成的构象未能捕捉这一特征。传统方法生成的构象中,苯环取向偏离实验确定的晶体构象,导致RMSD(均方根偏差)普遍超过2.00 Å。文章通过图1(原文图4-B)直观对比了A1O分子的晶体构象与传统方法生成构象的差异。
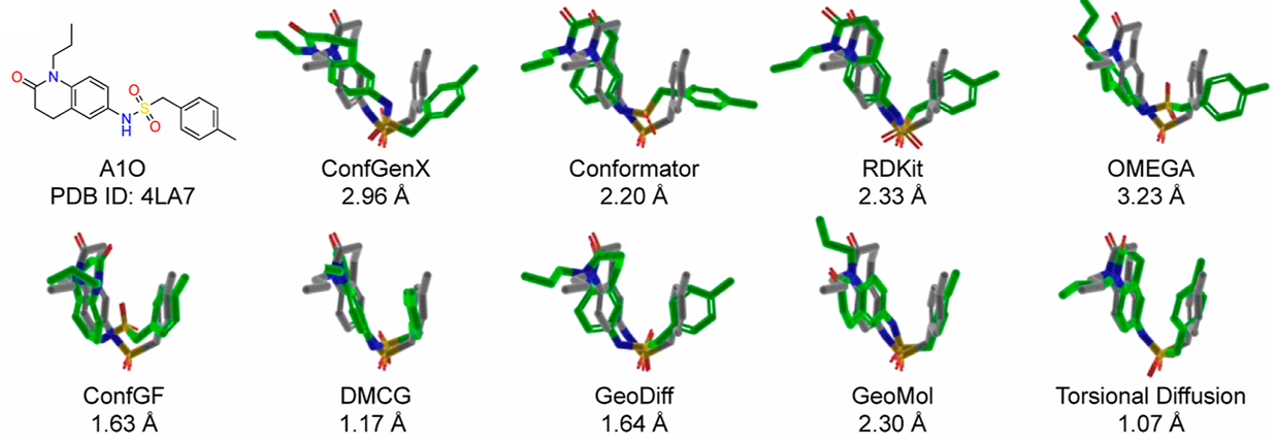
图1. 代表性分子A1O的生成构象与晶体构象对比。生成构象中的碳原子和晶体构象中的碳原子分别用绿色和灰色表示,所有氢原子均未显示。图片来自文献[1]。
如图1所示,传统方法生成的构象(绿色)与晶体构象(灰色)在苯环空间取向上存在显著差异,而人工智能模型生成的构象更接近实验观测状态。具体而言,传统方法倾向于生成舒展型构象,而非折叠型生物活性构象。Wang等人1从方法学角度解释了传统方法的局限性:
- 规则库的局限性:传统方法(如OMEGA、ConfGenX)依赖从晶体数据库(如CSD或PDB)推导的构象偏好(如扭转角、环几何),但A1O分子的π-π相互作用不属于常见规则范畴,导致预测偏差。
- 能量最小化偏向:传统方法倾向于生成热力学最稳定的构象(能量最低),而生物活性构象可能因蛋白环境而处于亚稳态。A1O分子的结合构象涉及熵效应对不利的取向,传统方法的力场优化无法模拟这种蛋白诱导的构象变化。
- 采样策略不足:传统方法基于距离几何或随机采样,对于柔性分子的构象空间覆盖不全面。A1O分子需精确的扭转角组合,传统方法在有限采样下易错过关键构象。
基于此,Wang等人1推测:”对于具有强分子内相互作用的生物活性构象预测,人工智能模型可能是更优选择。”
本文提出不同见解:传统方法的失效主因在于默认力场参数设置。为了平衡速度与精度,默认参数使用的变体力场通常忽略了分子内长程相互作用——这使得分子内强的相互作用被忽略。
以omega的searchFF选项为例2,3:
1 | -searchFF : Forcefield to be used for torsion driving |
该力场参数有多个选项2,3:
1 2 3 4 5 6 7 8 9 | Contents of parameter -searchFF
Aliases : -srchff
Type : omegaff
Allow list : false
Default : mmff94smod_noestat
Simple : false
Required : false
Legal values : mmff mmff_noestat mmff_trunc mmff_sheff mmff94s mmff94s_noestat mmff94s_trunc mmff94s_sheff mmff94smod mmff94smod_noestat mmff94smod_trunc mmff94smod_sheff sage sage_noestat sage_sheff
Brief : Forcefield to be used for torsion driving |
注意到:mmff94smod_noestat是searchFF选项的默认参数。根据说明书3对mmff94smod_noestat的描述,该力场变体包含除库仑相互作用之外的所有 MMFF94s-Mod 项。也就是说,在默认情况下,omega为了平衡计算速度与精度,刻意排除了分子内库仑相互作用项(即静电作用),导致长程非键相互作用被忽略。这可以解释为什么Wang等人1观察到使用传统方法不能重现A1O的生物活性构象。
Flare4的构象搜索工具Conformation Hunt(对应的命令行工具为XedeX5)是另一个采用类似策略的构象搜索方法,在默认条件下忽略分子内长程相互作用,如下图2所示。
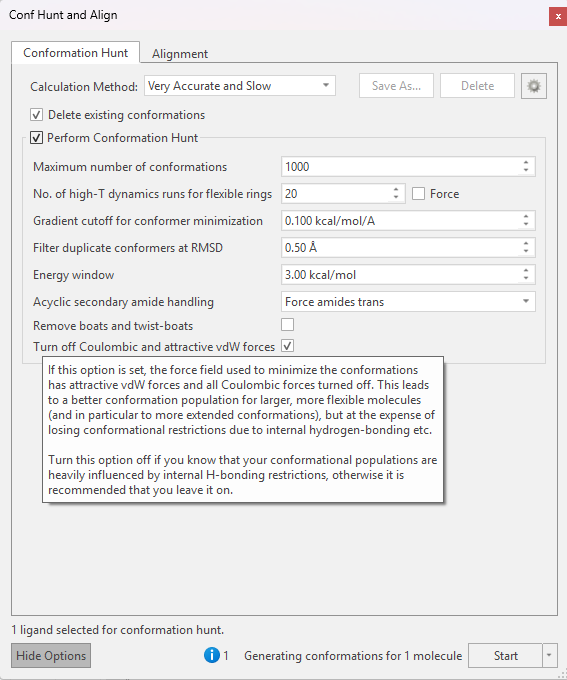
图2. Flare构象搜索参数4
请注意,Flare 构象搜索在默认情况下”Turn off Coulombic and attractive vdW forces“选项是被选中的,如表1所示。这意味着:如果设置此选项,用于最小化构象的力场将关闭吸引性范德华力和库仑力。这会提高对更大、更柔性分子的采样效率(特别是更扩展的构象),但代价是削弱分子内氢键等关键相互作用的描述能力。如果知道构象受分子内氢键等关键相互作用影响很大,则需要关闭此选项,否则建议保持开启。
在实践中,除了分子内经典的氢键之外,该选项还对其它分子内相互作用产生影响,比如O/N···S相互作用、C-H···O=C等非经典氢键相互作用、卤键、π-π以及Cation-π相互作用等。在最近的一个构象搜索案例中用FieldTemplater预测ROS1抑制剂的生物活性构象6,涉及分子内N···S相互作用,强调一定要关闭”Turn off Coulombic and attractive vdW forces“项以确保得到正确的生物活性构象。在10年前更早的一个案例中,分享了共晶结构PDB 1S63的配体构象搜索案例7,发现长程相互作用选项对重现分子内Cation-π相互作用的影响。
| Items | Normal | Very Accurate and Slow |
|---|---|---|
| Maximum number of conformations | 100 | 1000 |
| No. of high-T dynamics runs for flexible rings | 5 | 20 |
| Gradient cutoff for conformer minimization | 0.500 kcal/mol/Å | 0.500 kcal/mol/Å |
| Filter duplicate conformers at RMSD | 0.5Å | 0.5Å |
| Energy window | 6.00 kcal/mol | 3.00 kcal/mol |
| Acyclic secondary amide handling | Force amides trans | Force amides trans |
| Remove boats and twist-boats | No | No |
| Turn off Coulombic and attractive vdW forces | Yes | Yes |
以Flare构象搜索为例,在标准构象搜索模式(Normal mode,表1)下,是否启用Turn off Coulombic and attractive vdW forces选项对生成构象的准确性具有显著影响。如图3所示,当该选项被激活(即关闭静电和吸引性范德华作用)时,所获得的计算构象(图3中绿色)与生物活性构象(图3中黄色)在苯环取向上存在明显偏差,未能形成分子内“面对面”的π-π堆积相互作用。相反,当该选项被禁用(即保留静电及吸引性范德华长程作用)时,计算所得构象(图3中棕色)在苯环取向上与生物活性构象高度一致,成功重现了关键的分子内“面对面”的π-π相互作用模式,且重原子根均方偏差(RMSD)仅为1.26 Å,表明构象匹配度显著提升,并Wang等人1报道的人工智能方法(图1)精度相当。上述结果表明,保留长程静电与吸引性范德华相互作用对于精确捕获生物活性构象中的关键非共价相互作用具有重要意义。
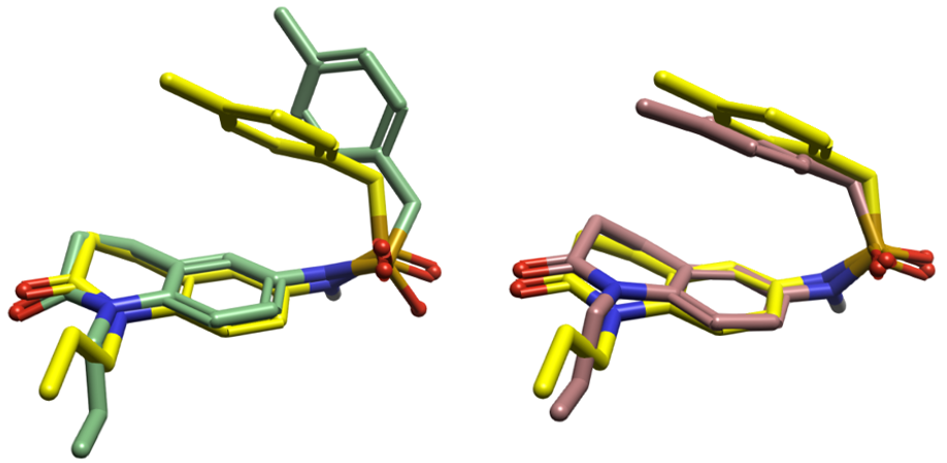
图3. A1O生物活性构象(黄色)与Flare 生成构象的比较,绿色:关闭静电与吸引性范德华长程相互作用的计算构象,棕色:打开静电与吸引性范德华长程相互作用的计算构象。
当然构象搜索还受到其它因素影响,比如重复构象的定义,能量窗大小的设置等影响。在标准模式下,如图3所示,在内酰胺N原子上取代基丙基的构象差异,可能源于构象覆盖度不足。在Flare的Conf Hunt非常精确模式下(Very Accurate but slow,表1),如图4所示,就正确地找到A1O的生物活性构象:黄色分子为PDB 4LA7共晶配体A1O的结合构象,绿色分子为Flare 搜索到的一个低能构象,两者之间的重原子RMSD = 0.96 Å,精度优于Wang等人1报道的所有人工智能方法(图1)。Omega在使用标准力场的情况下,也重现了A1O的结合构象。
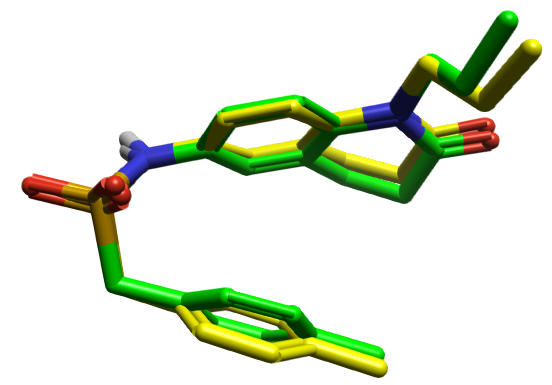
图4. A1O生物活性构象(黄色)与Flare 生成构象(绿色)的比较
综上,传统构象生成方法的表观”失效”,实质源于力场长程相互作用参数配置不当,而非方法学固有缺陷。合理调整参数可显著提升对强分子内相互作用体系的预测能力。
推荐阅读
《用XedeX进行构象搜索》:在这篇博客中,以PDB 1S35的共晶配体778为例,激活“静电与吸引性范德华”选项比关闭该选项可以更好地描述分子内阳离子-π相互作用,重现778的生物活性构象。
文献
- Wang, Z. et al. (2023) “Small-Molecule Conformer Generators: Evaluation of Traditional Methods and AI Models on High-Quality Data Sets,” Journal of Chemical Information and Modeling, 63(21). Available at: https://doi.org/10.1021/acs.jcim.3c01519.
- OMEGA 5.1.0.0: OpenEye, Cadence Molecular Sciences, Santa Fe, NM. http://www.eyesopen.com
- Omega Documnet. OpenEye, Cadence Molecular Sciences, Santa Fe, NM. https://docs.eyesopen.com/applications/omega
- Flare V10. Cresset. https://www.cresset-group.com/software/Flare
- XedTools. Cresset. https://cresset-group.com/software/xed-tools/
- 用FieldTemplater预测ROS1抑制剂的生物活性构象. 墨灵格的博客. Available at: http://blog.molcalx.com.cn/2025/10/17/prediction-of-bioactive-conformation-of-ros1-inhibitors.html
- 用XedeX进行构象搜索. 墨灵格的博客. Available at: http://blog.molcalx.com.cn/2025/11/16/conformation-search-performance-with-xedex.html


