
摘要:迈克尔(Michael)受体与半胱氨酸巯基加成是几种共价药物(例如afatinib、osimertinib、ibrutinib、neratinib和CC-292)的作用机制。已有文献报道了可逆的Michael受体,这些Michael受体的α-碳上具有另额外的吸电子基团。我们进行了密度泛函理论计算以确定为什么这些Michael受体的硫醇加成是可逆的。α-EWG基团稳定了阴离子过渡态和Michael加成反应的中间体,但较不直观,它会使中性的加成物不稳定。这使得逆反应(消除)更快且热力学更有利。为了使硫醇的加成可逆,Michael受体还必须在β-碳上具有合适的取代基,例如芳基或支链烷基。计算解释了这些结构元件如何促进可逆性以及它们调节结合亲和力与共价抑制剂停留时间的能力。
编译:肖高铿
原文:Krenske, E. H., Petter, R. C., & Houk, K. N. (2016). Kinetics and Thermodynamics of Reversible Thiol Additions to Mono- and Diactivated Michael Acceptors: Implications for the Design of Drugs That Bind Covalently to Cysteines. The Journal of Organic Chemistry, 81(23), 11726–11733. https://doi.org/10.1021/acs.joc.6b02188
前言
人们对理性设计那些能共价修饰生物靶标的药物越来越感兴趣[1]。传统上,出于安全考虑,基于结构的药物设计项目避免使用共价修饰剂。具有化学反应性的抑制剂发生脱靶相互作用的倾向与毒性和免疫反应的风险增加有关。但是,比起传统的非共价抑制剂,共价抑制剂可实现更高的结合亲和力[2]和更长的停留时间(residence times)[3],并且这些特性则可以变现为更低的剂量、给药频次和药物的全身性暴露。因此,许多制药公司启动了针对各种酶的共价抑制剂药物开发计划。
其中一个策略是“靶向共价抑制”,该策略已经产生了几个被FDA批准的药物[1]。在该策略中,共价的弹头与通过非共价相互作用识别靶蛋白结合位点的配体相捆绑。一旦配体对接进入结合位点,弹头就会与附近的氨基酸残基形成共价键。通过设计抑制剂以便共价键仅形成于保守性差的非催化残基,以此实现靶标选择性的最大化。
Michae受体的半胱氨酸巯基加成是靶向共价抑制剂设计的常见形式。靶向半胱氨酸的丙烯酰胺afatinib[4]和osimertinib[5]已获得FDA批准用于治疗非小细胞肺癌(NSCLC),而ibrutinib[6]已被批准用于治疗慢性淋巴细胞性白血病和套细胞淋巴瘤(SCHEME 1)。目前在临床试验中其它靶向半胱氨酸的Michael受体包括EGFR/ERBB抑制剂dacomitinib[7],CO-1686[8](NSCLC),neratinib[9](乳腺癌)和Bruton酪氨酸激酶(BTK)抑制剂[10]CC-292[11](血液癌)。所有这些抑制剂均不可逆地修饰其靶蛋白。
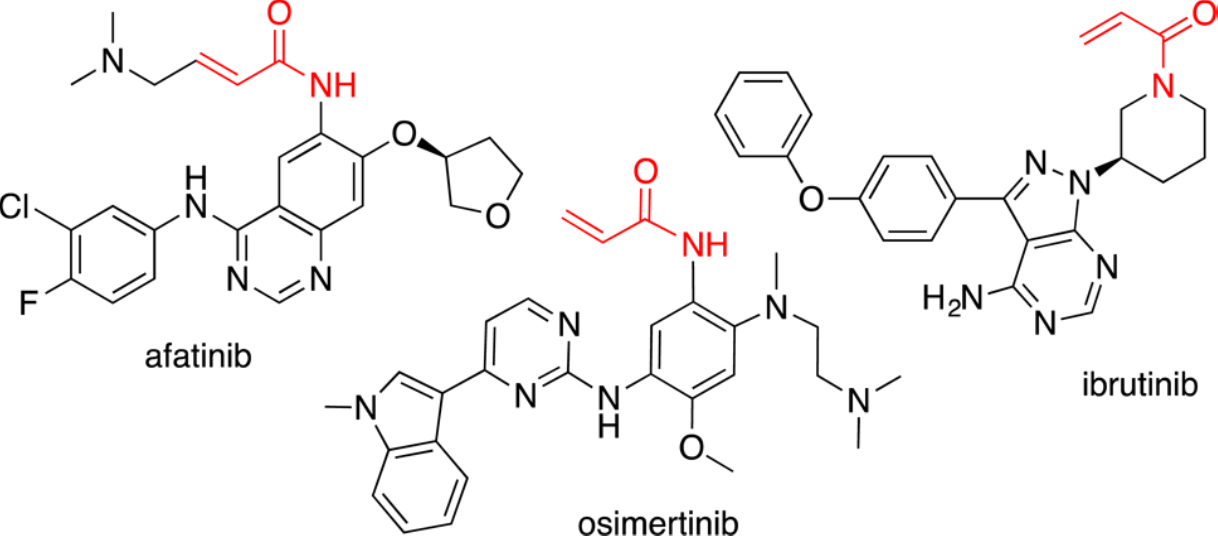
Scheme 1. 与蛋白Cys巯基不可逆结合的共价药物
最近发展的可逆共价抑制的概念,即其中的共价键可以很容易地断裂以释放抑制剂[3,12,13]。可逆键的形成带了开发与靶蛋白形成持久但不一定永久相互作用的抑制剂的前景。如果结合位点没有正确的三维结构(例如,如果蛋白质未折叠,如在天然蛋白质周转过程中发生的那样),或者如果抑制剂与脱靶的巯基结合,则这种结合将是短暂的而且危害较小。可逆共价抑制剂的潜在优势包括提高了安全边界和对慢性疾病的适用性。
Taunton及其同事率先设计了活化的Michael受体作为为可逆的半胱氨酸靶向抑制剂(Scheme 2)[12,14,15]。 而简单的Michael受体(如1和2)与巯基不可逆地反应生成可分离的加成物,而在α位上含有吸电子基团(EWG)的类似物(例如氰基取代的3)的巯基加成物不能分离出来并在稀释时还原成反应物。同样,受体4-7(其中3的羧酸被杂芳环取代)也显示出可逆的巯基加成反应[16]。这些α-EWG已用于设计几种激酶的可逆抑制剂[3,12,16-18]。例如, 与缺乏α-EWG的不可逆抑制剂10与11相比,α-氰基丙烯酸酯8和α-氰基丙烯酰胺9对p90核糖体蛋白S6激酶RSK2(RSK2-CTD)的C端激酶结构域的抑制作用高150倍。
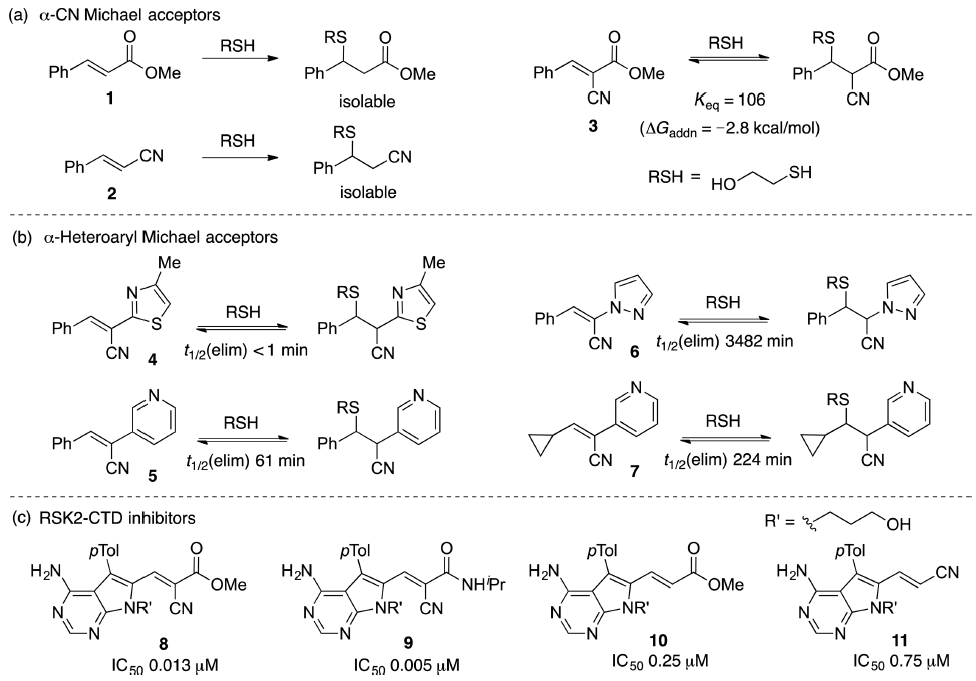
Scheme 2. (a) 吸电基团对硫醇Michael受体加成可逆性的影响[12],(b)α-杂芳基Michael受体和相应的β-巯基乙醇加成物的巯基消除半衰期[16],(c) RSK2-CTD12激酶的可逆与不可逆抑制剂
与这些观察结果相关的是抗炎药巴多索隆甲酯(bardoxolone methyl,CDDO-Me,12)[19],一种半合成齐墩果酸衍生物,已进入糖尿病患者慢性肾脏疾病的III期临床试验[1h,20]。对母体化合物Bardoxolone(13)的光谱分析表明巯基可逆地加成到α-氰基烯酮基团上[19a] 。Bardoxolone甲基的α-氰基烯酮部分可逆地修饰特定的蛋白质半胱氨酸残基,据认为可触发Keap1-Nrf2通路的激活并抑制NF-κB,导致观察到抗炎活性。
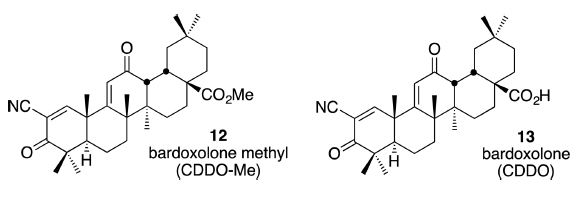
尚不清楚为什么α-EWG会使巯基-迈克尔受体的加成是可逆的。Taunton指出,α-EWG使巯基加合物更加酸性,稳定了碱催化消除过程中的中间体共轭碱(烯醇化物或相关阴离子)[12,16]。然而,α-EWG也能活化硫醇基阴离子(thiolate anion)与Michael受体的反应,这对C-S键的形成更有利。我们研究了这些效应对各种取代Michael受体的速率与平衡的影响。
本文报告了一项理论研究,阐述了Michael受体上的取代基如何影响巯基加成的动力学和热力学从而导致可逆性。密度泛函理论计算得出了出乎意料的结论,即α-EWG不仅降低了巯基加成或消除的能垒,而且使加成在热力学上的有利性较小。除α-EWG外,β-碳上的取代基对整体能量学也至关重要,是可逆抑制剂设计的同等重要因素。
结果与讨论
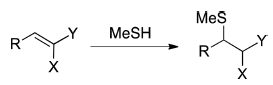
一系列Michael受体(表1)与硫醇的加成采用密度泛函(DFT)进行计算[21]。最初,我们通过比较将β-巯基乙醇加成到α-氰基取代的Michael受体3和14-18上的ΔG的计算值和实验值[12],评估了几种DFT方法的性能(表1A)。在计算中,将β-巯基乙醇建模为MeSH[2]。我们评估了各种泛函(B3LYP,M06-2X和ωB97X-D)、基组、计算熵的方法和溶剂化模型。支持信息中提供了完整的详细信息。在所检查的方法中,与M06-2X/6-311+G(d,p)结合CPCM连续溶剂模型进行的计算获得了与实验最接近的结果。下面的讨论将参考用这种理论方法获得的结果,支持信息中讨论了其他理论水平的计算结果。
表1. 水中迈克尔受体上硫醇加成物的热力学计算a
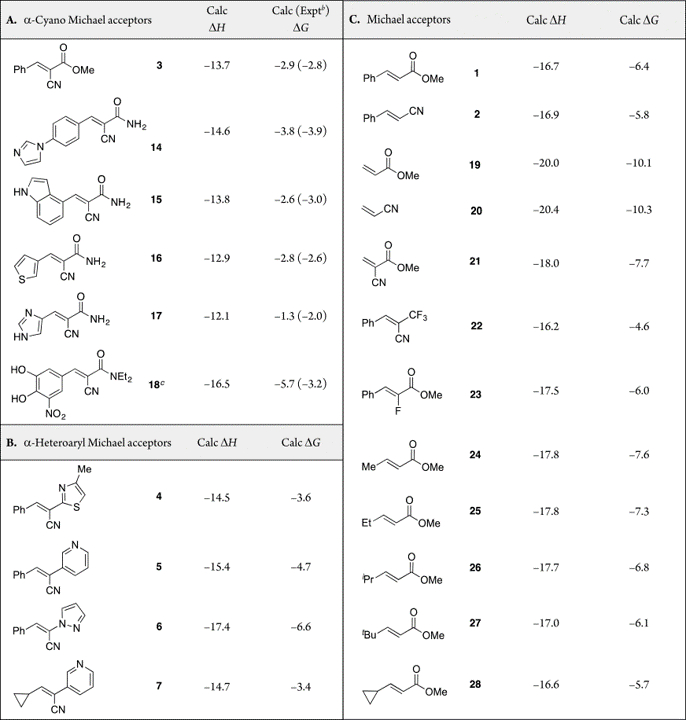
a:ΔH和ΔG的单位为kcal/mol,在M06-2X/6-311+G(d,p)理论水平计算,以CPCM溶剂化模型考虑水的效应。b:β-巯基乙醇的Michael加成ΔG(1-2%DMSO的PBS)数据来源自文献12;NEt2用NMe2模拟。
在大多数情况下,计算出的ΔG值与实验值ΔG值之间的一致性非常好,在0.1-0.7 kcal/mol之内。恩他卡朋(18)是例外。预测的ΔG值比实验值负2.5 kcal/mol,这可能是由于在实验条件下(PBS, 1-2%DMSO)18的硝基邻苯二酚基团被部分离子化的缘故,该计算未建模。总的来说,这些计算正确地预测了ΔG的绝对值和α-氰基取代的Michael受体的相对反应性的一般趋势。
表1B,C显示了MeSH与一系列Michael受体加成反应的计算结果。表1B精选了一些α-杂芳基丙烯腈。 Taunton[16]报道了从这些受体的加成物中消除β-巯基乙醇的半衰期,并显示在图2b中。表1C给出了一系列迈克尔受体模型,经计算可用于分析取代基效应。
从表1中可以清楚地看到两个观察结果。首先,MeSH加成到α-氰基丙烯酸酯(例如3和21)比起加成到相应的母体化合物丙烯酸酯(1和19)或丙烯腈(2和20)在热力学上有利,低约3kcal/mol。加成到α-氰基衍生物中的热力学驱动力较低,这出乎我们的意料[23]。这表明可逆性不仅取决于加成物的酸性。的确,较小的热力学驱动力是3与硫醇加成物在稀释时会被消除的根本原因[12],而与α-未取代的类似物1和2加成实际上是“不可逆的”。
硫醇与1或2的加成平衡常数约为K=104(ΔG=-6kcal/mol),而与3的加成平衡常数K=102(ΔG= -3 kcal/mol)。化合物3反应的平衡位置比1或2的反应对浓度更敏感。例如,在100mM硫醇和100μM 1的起始溶液中,在平衡状态下100%的1都转化为硫醇加成物。如果将平衡混合物稀释10倍,则只有0.2%的加成物还原为1和游离硫醇。相反,在相同浓度下3与硫醇的反应将使93%转化为加成物,稀释10倍后,39%的加成物将还原为3和游离硫醇。为了通过实验检测到3与巯基加成的可逆性,加成和消除的动力学必须快速。但是,如果氰基基团不影响ΔG的话,那么即使势垒很低,3的加成物也会发生极少量的硫醇消除。
MeSH加成至α-杂芳基取代的丙烯腈4-7的ΔG值在-3.4至-6.6 kcal/mol之间。这些值介于MeSH加成至氰基丙烯酸酯3的ΔG值(-3 kcal/mol)与加成至1或2的ΔG值(-6 kcal/mol)之间,吡唑基衍生物6是个例外,它的ΔG= -6.6kcal/mol。从实验上看,硫醇与化合物4-7的加成是可逆的,消除半衰期为秒(4)、分钟(5)、小时(7)或天(6)[16]。预测α位的σ-受体基团(CF3)也对加成不太有利,如化合物22所示,其中α-CF3基团使ΔG升高的量与化合物5的3-吡啶基的升高量相当(ΔG= -4.6 kcal/ mol)。C-α(23)处的氟取代基的作用较小,与1相比,ΔG提高了0.4 kcal/mol。已有人探索将α-CF3-和α-F-取代的丙烯酰胺作为设计耐药EGFR突变体的可逆共价抑制剂的构建体[24,25]
表1中的第二个重要发现是,α-EWG本身不足以使硫醇的加成可逆。 Michael受体β-碳上取代基起着同样重要的作用。MeSH加成到没有β取代基的Michael受体(19-21)的ΔG值比加成到相应的β-苯基取代的受体(1-3)的ΔG低约(更加负值)4-5 kcal/mol,并预测加成至未取代的Michael受体应是高效不可逆[26,27]。因此,化学修饰Michael受体,使其与硫醇可逆反应的能力不仅取决于α-EWG,而且还取决于是否存在合适的 C-β取代基。
最早的可逆半胱氨酸靶向抑制剂(例如Scheme 2的8和9)在β位置含有杂芳基取代基,但最近的工作[3,16]在此位置使用了支链烷基。我们先用6-31 +G(d)基组进行了硫醇加成计算随后再用6-311+ G(d, p)基组进行单点能计算。总之,预测将MeSH加成至12得到33,其ΔG=-2.5 kcal/mol。这个较小负值的ΔG与实验相符,该实验表明向Bardoxolone加成硫醇是可逆的[19a]。有趣的是,预测烯醇32的能量几乎与酮33相同(-2.2 kcal/mol)。该结果也与原始实验报告一致,在该实验中,基于与已知烯醇34的光谱相似性,将巴多索隆在水溶液或DMSO中的硫醇加成物的结构指认为烯醇式而不是酮式[19a,28]。烯醇32相对较高的稳定性并不是α-氰基Michael受体加成的一般特性:作为比较,3的MeSH加成物作为烯醇比作为酮的稳定性低15kcal/mol[29]。
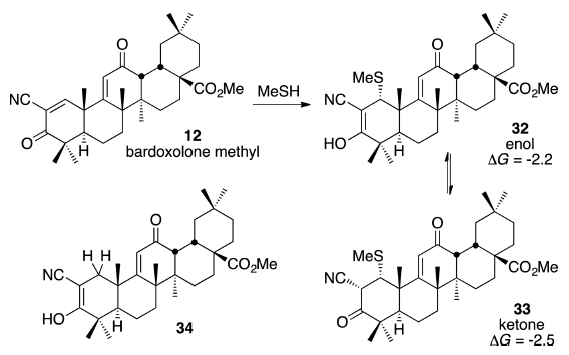
图1. MeSH的巴多索隆甲基加成: 在M06-2X/6-311+G(d,p)//M06-2X/6-31+G(d)理论水平 ,用CPCM溶剂化模型考虑水的溶剂效应(ΔG的单位是kcal/mol)
为了确定α-EWG和β-Ph基团如何影响硫醇加成的动力学,我们计算了碱催化MeSH与Michael受体1-3、19的加成自由能曲线(图2)。Michael加成反应的第一步是使巯基去质子化得到硫醇根阴离子。硫醇盐与Michael受体加成得到烯醇盐(或相关阴离子)中间体是速率决定的[30]。许多通用或专一的碱催化剂可在缓冲溶液中或细胞内使巯基去质子;我们使用DBU作为模型碱,因为这可以在能量尺度上方便地比较离子和中性反应。在水溶液中,碱催化剂的选择会影响硫醇盐阴离子和烯醇盐(或类似烯醇盐)中间体的稳定性,这是由于pKa在相关质子传递平衡上的作用,即[BH +] [MeS-]和[BH +] [Int]的相对能量[31]。然而,碱对巯基加成的整体热力学没有影响,也没有影响游离硫醇根阴离子与Michael受体加成的能垒。
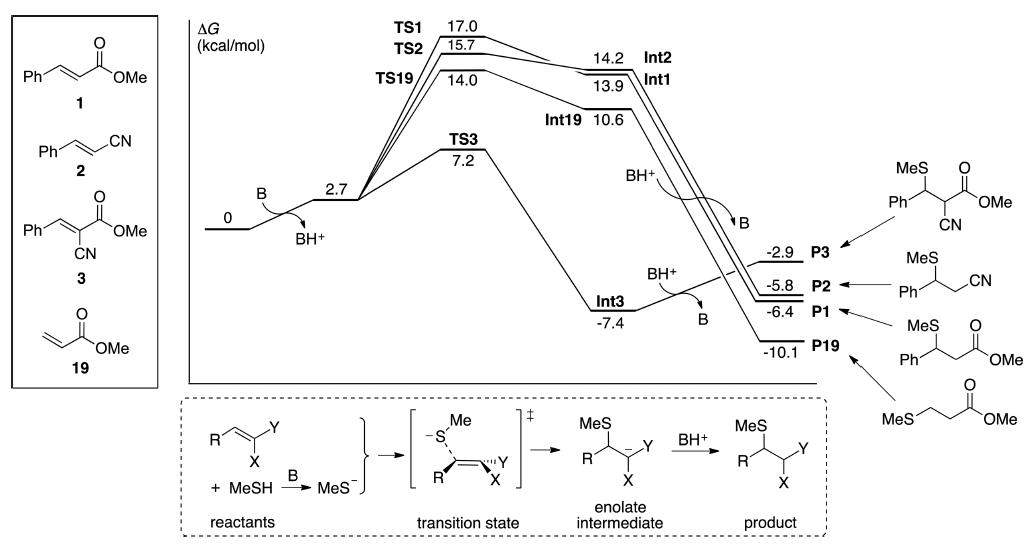
图2. 水溶液中MeSH加成到Michael受体1-3和19分别生成产物P1-P3与P9的自由能曲线,计算在M06-2X/6-311+ G(d,p)理论水平下进行,并采用CPCM溶剂化模型考虑水的效应。 DBU作为碱催化剂的模型分子(B)。
MeSH与化合物1和2上的不可逆加成具有相似的自由能特性,P1和P2分别为ΔG=-6.4和-5.8 kcal/mol,TS1和TS2的ΔG⧧分别为17.0和15.7 kcal/mol。中间体(Int1和Int2)位于决定速率的过渡态下面1-3 kcal/mol。相比之下,MeSH加成到α-氰基丙烯酸酯3的能量表面具有质的不同。过渡态(TS3)的能量比TS1或TS2低约9 kcal/mol(ΔG⧧= 7.2 kcal/mol),烯醇酯(enolate)(Int3)比Int1或Int2稳定约21 kcal/mol(-7.4 kcal/mol)。在双取代的烯醇酯中(Int3),每个EWG的负电荷离域能力几乎与单取代的类似物相同。在Int3中,CO2Me基团(-0.60 e)和CN基团(-1.15 e)上的Mulliken偏电荷(partial charge)类似于Int1中的CO2Me基团(-0.63 e)和Int2中的CN基团(-1.37 e) 。这对应于加成能力基本相当的两个负电荷离域EWG,这可以解释Int3为什么有强大的稳定性。计算证实了Taunton等人的建议,即α-EWG引起加成物酸度显着增加[12]。然而,酸度的增加既是由于烯醇物的稳定化结果,又在较小程度上是由于加成物不稳定的结果。即,尽管烯醇式Int3比Int1或Int2稳定21kcal/mol,但是加成物P3比P1或P2稳定性差了约3kcal/mol。α-EWG对产物稳定性的影响不同于其对反应物、TS和中间体稳定性的影响。
尽管α-EWG对过渡态和中间体可观的稳定化作用是加成和消除速率加速的原因,但硫醇与α-EWG底物加成的可逆性(即消除)这一事实源自较快的反应速度和整体较小的ΔG,后者与动力学无关。
图2还显示了β-芳基如何影响硫醇加成的动力学和热力学。β-Ph-取代的丙烯酸酯1与未取代的类似物19的比较表明,C-β苯基抬高了硫醇加成能垒3kcal/mol,并且在总体上使加成产生了4kcal/mol的不利。因此,与P19相比,P1的逆反应能垒(硫醇消除)更低,这对应于Ph取代的衍生物更快地发生消除。MeS-与β-烷基取代的Michael受体24-28(请参阅支持信息)加成的计算表明,Me、Et和iPr衍生物加成的能垒与Ph取代1的能垒相似,而总ΔG值更负。因此,硫醇加成到这些受体的速率与1相似,但是从加成物的硫醇消除速度将较慢。 tBu或环丙基会使能垒增加1-2 kcal/mol;这将降低加成速度,但对消除速度的影响较小。

图3.等键方程量化了α-CN和β-Ph取代基对烷烃和烯烃稳定性的影响。ΔH值(kcal / mol) 用CBS-QB3计算。
为什么双活化Michael受体的加成在热力学较不利呢?图3方程1-6中的等键反应( isodesmic reaction)定量考察了α-CN和β-Ph对不饱和与饱和物种稳定性的影响。 在这些方程中,高精度的CBS-QB3用来计算气相ΔH值,从而可以在没有熵或溶剂化作用的情况下分析取代基的固有作用。方程1表明氰基优选连接至sp2烯烃碳上而不是sp3烷烃碳上。方程2表明,偕双氰基更偏好sp2环境,但该优先级高于单个氰基。方程3表明Ph基团与sp2烯烃碳连接的优先性更大,约为单个CN基团的4倍。当上述烯烃带有α-CN基团(方程4)时,该优先性又进一步增加了1 kcal/mol。
单个CN或Ph基团优先连接到sp2碳而不是sp3碳是由于通过共轭作用使烯烃稳定。对于双-CN取代,烯烃的共轭起了作用,但对sp2环境的偏好主要反映了饱和产物的去稳定化作用。图3等式5和6的量化分析结果表明,虽然双氰基对饱和碳(eq5)或不饱和碳(eq 6)的偕双取代与参比单取代物相比是去稳定化的,但是对饱和碳的双氰基取代引起的去稳定大于1 kcal/mol。第一个EWG使饱和碳更正,而第二个EWG的添加是不利的。在烯烃中,由于第一个氰基引入的引起额外正电荷通过π键的极化稳定到一定程度(会被β-芳基会进一步增强),因此去稳定作用的程度会略微小些。两者合计,图3中的等键反应表明,巯基加成反应对带有α-EWG和β-芳基的Michael受体的可逆性源于反应物稳定化和产物去稳定化的结合,最大的单取代基效应是β-芳基。
结论
理论计算表明,硫醇Michael加成的反应性和热力学之间没有普适性的关系。硫醇加成物形成的可逆程度同时取决于这两个因素。Michael受体上的α-EWG降低了硫醇加成的能垒,但也使加成在热力学上不太有利。α-EWG与合适的C-β取代基(例如芳基或支链烷基)一起产生快速可逆的、弱能量的硫醇加成。加成物、硫醇和沿着加成或消除路径使这两种物质去质子化的碱的酸度并不直接影响热力学,而是影响反应速率。该反应速率最终决定了在化学或生物学相关时间尺度上将发生多大程度的硫醇消除。
在化学实验中,硫醇加成的可逆性程度由加成和消除的ΔG与ΔG⧧值定义。在共价药物设计中,这些特性只是更为复杂图片中一小片。抑制剂的结合亲和力和on/off速率还取决于局部pH和离子强度、半胱氨酸残基和催化碱基的pKa、硫醇加成物的构象、α-质子的可及性、浓度波动、重要的是抑制剂与靶标结合位点之间的非共价相互作用。[3,12] 不要期待溶液中反应的计算会与抑制剂的效果或停留时间完美相关,但它们确实揭示了可逆性弹头反应性的重要基本特征,并证明可逆的硫醇反应性是可以通过计算机方法来预测的。我们的计算考虑了一系列不同类别的可逆共价弹头,这些弹头对多种治疗靶标的新型抑制剂的设计颇有前景。
理论计算
密度泛函理论计算采用Gaussian 09在M06-2X/6-311+G(d,p)理论水平进行计算, 水溶液使用CPCM隐式溶剂模型模拟,并使用了超精细积分网格。谐波振动频率计算不仅用来验证驻点是最小状态还是过渡状态,而且还给出了无标度的零点能量和热校正。吉布斯自由能是在1mol/L的浓度、25°C的标准状态下计算的。选择M06-2X/6-311+G(d, p)来模拟硫醇的加成是基于验证研究。在验证研究中我们评估了多种泛函(B3LYP、M06-2X和ωB97X-D)、基组、计算熵的方法和溶剂模型。这些计算的详细信息在支持信息中给出。对于图3中的等键反应,采用高精度的CBS-QB3方法进行计算。


