
摘要:在《Gaussian教程–ECD计算》一文已经详细描述过如何预测ECD图谱,但是还有许多人要求更详细的说明,尤其是如何生成Excel报告。本文以Dexmedetomidine的ECD图谱预测为例,详细说明了如何用Excel进行后期的数据处理,包括:ECD图谱的生成,玻尔兹曼权重的计算,加权平均图谱的生成等等。还演示了如何用SpecDis生成玻尔兹曼加权平均图谱。
肖高铿/2020-01-01
前言
在《Gaussian教程–ECD计算》一文已经详细描述过如何预测ECD图谱,但是还有许多人要求更详细的说明,尤其是用Excel生成报告。本文以Dexmedetomidine的ECD图谱预测为例,详细说明了用Excel进行数据处理的完整过程。
算例化合物
化合物结构
Dexmedetomidine
SMILES代码
1 | C[C@@H](c1cccc(C)c1C)C2=CNC=N2 |
准备初始的3D结构
可以用Chem3D绘制算例化合,再用MMFF94优化,保存为MDL MOL FILE格式文件;或者用GaussView 6绘制结构,clean up后,保存为MDL MolFile格式文件(注意:不是MDL MolFile 3000)。我已经准备好了一个文件:Dexmedetomidine.mol。可以点击下载使用。
构象搜索
我们提供了molconflex.MPI脚本程序来调用CONFLEX进行构象搜索。该命令以MDL MOL FILE格式的3D结构文件为输入文件,默认采用12个核心进行构象搜索计算。因此,此处的输入文件(也就是命令的参数)为Dexmedetomidine.mol。
构象搜索的命令如下:
1 | molconflex.MPI Dexmedetomidine.mol |
请耐心等待构象搜索完毕,标记是命令行提示符又回来了。CONFLEX生成的全部文件都保存在一个以conflex为前缀的文件夹里,此处为conflex.Dexmedetomidine。用ls命令查看该文件夹的内容:
1 2 3 4 5 | ls conflex.Dexmedetomidine Dexmedetomidine.bso Dexmedetomidine.ls1 Dexmedetomidine.mol Dexmedetomidine.fxf Dexmedetomidine.ls2 Dexmedetomidine.sdf Dexmedetomidine.fxo Dexmedetomidine.ls3 Dexmedetomidine.ini Dexmedetomidine.ls4 |
该文件夹有两个文件很重要:Dexmedetomidine.ls4与Dexmedetomidine.sdf。ls4文件给出了每个构象的构象分布,用less命令可以查看:
1 | less conflex.Dexmedetomidine/Dexmedetomidine.ls4 |
我们关心下面的这个部分:
1 2 3 4 5 6 7 8 9 10 11 | ======================================================================================================================================== CONF. SYM. POINT NO. NO. ID HE TSE GE DGE GPOP SE DSE EPOP CHIRAL NUM. GROUP INIT. REOPT. NEG. ---------------------------------------------------------------------------------------------------------------------------------------- 1 00000001 221.5036 35.6694 185.8342 0.0000 45.1652 48.8457 0.0000 47.0162 T 1 C1 T F 0 2 00000006 222.0239 35.9992 186.0247 0.1904 32.7496 49.2567 0.4110 23.4962 T 1 C1 T F 0 3 00000004 221.8718 35.5998 186.2719 0.4377 21.5750 49.1525 0.3068 28.0111 T 1 C1 T F 0 4 00000002 223.9685 35.1435 188.8250 2.9908 0.2901 51.2498 2.4041 0.8129 T 1 C1 T F 0 5 00000003 224.0975 35.1066 188.9909 3.1567 0.2193 51.3723 2.5266 0.6611 T 1 C1 T F 0 6 00000005 227.4734 35.1810 192.2923 6.4581 0.0008 54.6699 5.8242 0.0025 T 1 C1 T F 0 ---------------------------------------------------------------------------------------------------------------------------------------- |
除了在linux命令行可以查看外,你还可以将这部分内容复制到Excel表格进行分析。我们关心CONF. ID列,GPOP与EPOP列。其中GPOP与EPOP分别根据吉布斯自由能与张力能计算的玻尔兹曼分布。很明显,无论是GPOP还是EPOP,构象00000001,00000006与00000004占据了主要的构象空间,玻尔兹曼分布分别为:99.49%与98.53%。
而Dexmedetomidine.sdf文件则保存了全部搜索到的构象。构象在该文件中是按EPOP从高到低排序,我们可以输出该文件中CONF ID的顺序,用gawk命令就可以:
1 | gawk '/CONFORMER_ID/{getline;print}' conflex.Dexmedetomidine/Dexmedetomidine.sdf |
结果如下:
1 2 3 4 5 6 | 00000001 00000004 00000006 00000002 00000003 00000005 |
很明显,CONFORMER_ID是按EPOP从高到低排序的。了解这一点对于我们挑选化合物的构象很重要。接下来,我们就要将Dexmedetomidine.sdf中的每一个构象都提取出来,脚本molsplit可以方便的拆分结构:
1 | molsplit conflex.Dexmedetomidine/Dexmedetomidine.sdf |
结果提示:
1 2 3 | There is 6 conformer(s) in the conflex.Dexmedetomidine/Dexmedetomidine.sdf. 6 molecules converted 6 files output. The first is CONF_1.mol2 |
molsplit将6个构象从sdf文件中提取出来,按EPOP从高到低依次保存为TRIPOS MOL2格式文件,它们的命名规则为CONF_n.mol2, 其中n为从1开始的整数。本算例中生成了CONF_1.mol2, CONF_2.mol2… CONF_6.mol2等6个文件,分别与ls4的CONF. ID 00000001, 00000004, 00000006, 00000002, 00000003与00000005对应。
DFT结构优化与TD-DFT激发态计算
脚本程序m2ecd用于将mol2或sdf格式的3d分子结构转化为Gaussian输入文件,并进行DFT结构优化与频率计算以及TD-DFT激发态计算。以CONF ID为00000001的CONF_1.mol2文件为例,命令如下:
1 2 3 4 5 6 7 8 | [gkxiao@master Dexmedetomidine]$ m2ecd CONF_1.mol2 Please select a solvent by type a number: 1-Methanol; 2-Acetonitrile; 3-Chloroform 4-others: 1 The solution is Methanol Please select method and basis set by type a number: 1-APFD/6-311+g(2d,p); 2-B3LYP/6-311+g(2d,p);3-other: 1 1 molecule converted Submitted batch job 2962372 |
根据提示,选择您需要的溶剂进行DFT计算与TD-DFT计算。如果你所需要的溶剂没被列出,则选择4,并根据Gaussian在线手册SCRF关键词选择合适的溶剂;同时还要指定计算的方法与基组。m2ecd根据分子的电荷自行设定电荷与自选多态性,请务必对生成的Gaussian文件进行检查,确保结构、电荷与自旋多态性都是对的。
当你的溶剂、计算方法与基组选定之后,m2ecd会自动提交计算作业。默认采用Torque的PBS进行作业管理,你可以在后续通过Torque进一步用管理提交的作业。注意到第8行2962372是作业管理系统的作业号,记住这个号码很重要。
m2ecd为每个构象生成了4个文件,以CONF_1.mol2为例,分别生成了CONF_1_min.com, CONF_1_ecd.com, CONF_1_min.pbs与CONF_1_ecd.pbs四个文件。其中.com文件是gaussian计算的输入文件,.pbs是作业管理系统的脚本文件。gaussian计算就是通过管理系统来进行作业的提交、排队、挂起(暂停)、取消以及查询状态等操作。
PBS常用命令,可以bing或百度搜索Torque说明书或教程进一步了解,这里介绍少数几个命令:
1 2 3 | qstat -f 2962372 #查询作业号为2962372的作业状态 qdel 2962372 #取消作业号为2962372的计算作业 qsub CONF_1_min.pbs #提交CONF_1_min.pbs作业 |
你可以在m2ecd生成作业脚本的基础上,根据需要修改.com或.pbs文件,然后再提交作业。
作业计算完毕,每个构象会生成四个文件:
- _min.log文件
- _ecd.log文件
- _min.chk文件
- _ecd.chk文件
- .heat文件
- .cd.bil文件
_min.log是Gaussian的DFT优化与频率计算的结果文件,我们需要从中提取构象的自由能以计算玻尔兹曼权重。
_ecd.log是Gaussian的TD-DFT激发态T计算的结果文件,用于生成ECD图谱用。
_min.chk是Gaussian的DFT结构优化与频率计算的检查点文件。
_ecd.chk是Gaussian的TD-DFT进行激发态计算的检查点文件。
从2020年版本开始的m2ecd会提取频率计算的构象自由能用于计算玻尔兹曼加权。
从2020年版本开始的m2ecd会自动提取ECD图谱生成.cd.bil文件。与上面的.heat文件放在同一个目录下,可以直接用Specdis生成玻尔兹曼加权平均图谱(见视频演示:用SpecDis绘制图谱)。
此外,PBS作业管理系统也会生成两个日志文件:_PBS.err与_PBS.log。先不用管它们,对分析异常有帮助。
单个构象的ECD图谱绘制
跟据_ecd.log文件生成ECD图谱的具体操作步骤之前在《Gaussian教程–ECD计算》已经详细描述过,先用GaussView 6生成图谱并保存为文本格式的图谱文件,再用Excel等其它工具进一步处理。这里,我用Excel绘制了ECD图谱,详见附件:ECD_spectra.xlsx。
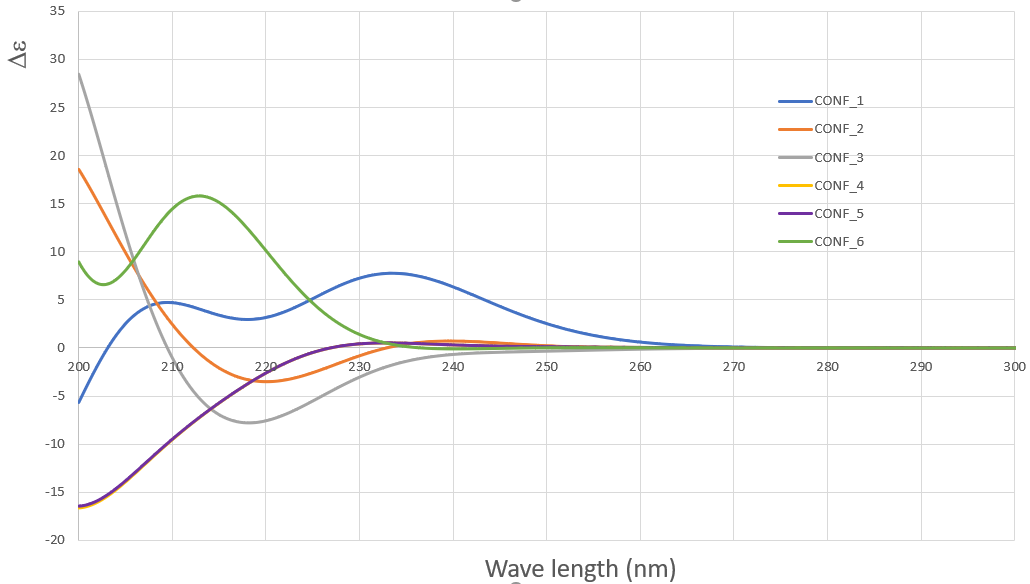
图2. 六个构象的ECD图谱各不相同(波长取值范围200-300nm)
玻尔兹曼权重计算
玻尔兹曼权重的计算方法在Gaussian教程 | NMR图谱与耦合常数的计算中已经详细描述过。简单地讲,就是从每个构象的_min.log文件里寻找”Sum of electronic and thermal Free Energies”行的能量值进行权重计算,结果如表1所示。
表1. 根据DFT优化与频率计算结果计算玻尔兹曼权重
| CONFORMER | Ei(Hatree) | ΔEi (KJ) | -ΔEi/RT | qi=e-ΔEi/RT | Weight |
|---|---|---|---|---|---|
| CONF_1 | -163.992870 | 3.890991 | -1.57 | 0.208 | 0.096 |
| CONF_2 | -613.993685 | 1.751209 | -0.706 | 0.493 | 0.229 |
| CONF_3 | -613.994352 | 0 | 0 | 1 | 0.464 |
| CONF_4 | -613.992396 | 5.135478 | -2.072 | 0.126 | 0.058 |
| CONF_5 | -613.992393 | 5.143355 | -2.075 | 0.126 | 0.058 |
| CONF_6 | -613.992853 | 3.935625 | -1.59 | 0.204 | 0.095 |
| ∑qi= | 2.16 |
其中,权重Weight=qi/∑qi
具体的计算过程详见附件:ECD_spectra.xlsx。
由表1可知,根据DFT计算的权重与MMFF94s力场计算的权重完全不同:根据MMFF94S力场的EPOP,有3个构象的空间分布不到1%,而DFT计算结果表明这六个构象都占据约5%以上的构象空间!一般用DFT方法计算的权重来对多个图谱进行“混合”绘制最终的图谱。
加权平均ECD图谱绘制
用GaussView6很容易实现对多个图谱按权重“混合”得到加权平均的图谱,详见《Gaussian教程–ECD计算》视频。也可以用Excel处理,在表单上每个构象图谱的Epsilon列乘以权重,再加和就得到新的图谱(图2),加权图谱生成过程详细见附件:ECD_spectra.xlsx。
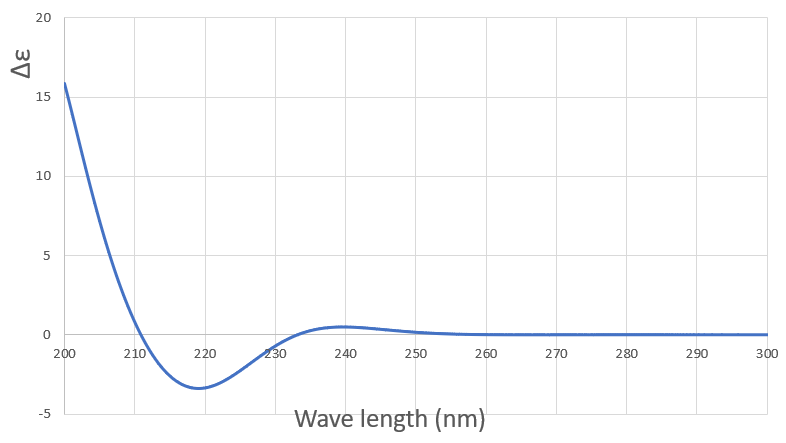
图2. 加权平均图谱
计算图谱与实验图谱的比较
将实验图谱与预测图谱都读入到同一个excel里,对计算图谱进行必要平移与缩放。百度会找到很多平移与缩放的教程,这里就不再重复说明了。
视频演示:用SpecDis绘制图谱
准备工作
SpecDis下载:https://specdis-software.jimdofree.com
演示材料下载:Spectra.zip
在演示材料里包含了同名的(1).heat后缀文件,该文件只有1行内容,为化合物构象自由能,单位为kcal/mol;(2).bil后缀文件,该文件是计算的ECD图谱,包含两列,其中一列为波长,另一列为Rotatory Strengths。
其中Heat文件用Specdis | Tools | extract heat读取对应的优化与频率计算结果文件(_min.log)生成。也可以手工生成,该文件仅包含一行以kcal/mol为单位的能量值。bil文件用Specdis | Tools | extract spectra读取对应的TD-DFT计算结果文件(_ecd.log)生成。bil文件可以手工一个一个生成。详细方法见SpecDis说明书。
注:2020年新版的m2ecd会自动提取ECD图谱生成.bil文件以及自动生成.heat文件,可以直接用SpecDis生成Boltzmann加权平均图谱。
单构象的图谱生成
Specdis | File | Open | 打开_ecd.log文件。生成的图谱可以保存为.xy的数据点格式或.bil文件。其中.bil文件在后面的玻尔兹曼加权图谱生成中要用到。
玻尔兹曼加权平均图谱
需要预先准备每个构象同名的.bil与.heat文件,保存与同一个文件夹里。这里我们准备了演示文件spectra文件夹,包含了6个构象的.bil与.heat文件。视频演示了如何用Specdis | Tools | sum spectra 生成Boltzmann加权图谱。
结构确证软硬件件解决方案
广州市墨灵格信息科技有限公司与广州五舟科技股份有限公司合作提供软硬件一体化的方案:将构象搜索、量子化学计算、结果分析与硬件集成在一起,让您尽快上手进行计算。



