
摘要:在不使用动物测试的情况下评估新化学品的安全性是一项非常重要的任务。 AMES试验是一种广泛使用的致突变性生物测定方法,对于动物源性试剂来说是个昂贵且浪费的过程。传统的AMES预测方法往往基于化学结构类型,该方法可能导致假阳性预测。化学结构分类的方法也忽略了与DNA反应性有关的内在化学本质。本研究用一个模型核酸碱基计算了30个1,4-Michael受体的活化能和HOMO/LUMO能量,并进一步用于预测这些化合物的AMES测试结果。所提出的模型建立在现有工作的基础上,并使用密度泛函理论过渡态模型研究了基本的毒物—靶标相互作用。结果表明,活化能小于20.7kcal/mol,ELUMO小于-1.85eV的Michael受体很可能就是DNA的直接诱变剂。
编译:肖高铿/2019-12-11
原文:Townsend, P. A.; Grayson, M. N. Density Functional Theory Transition-State Modeling for the Prediction of Ames Mutagenicity in 1,4 Michael Acceptors. J. Chem. Inf. Model. 2019, acs.jcim.9b00966. doi:10.1021/acs.jcim.9b00966.
前言
绿色化学等全球框架应处于现代化学研究的前沿。为了帮助实现这一思想,Anastas和Warner提出了绿色化学12原则作为开发绿色化学工艺和产品的基本指南。[1]计算毒理学领域涉及使用定性和定量数据来开发模型,以改善我们对化学物质的毒理学风险的理解。[2,3]这与绿色化学的第4个原则直接吻合:设计尽可能安全的化学品并降低毒性。此外,计算毒理学在试图减少动物实验的广泛使用方面发挥着关键作用——这是全世界人都非常关注的伦理问题。[4]尽管癌症的治疗方法不断改进,但致癌性仍然是开发新化学品时非常关注的毒理学终点。突变,即产生基因突变的过程,是许多癌症发生的基本过程。因此,了解致突变性的化学机理对于生产毒性较小的化学品至关重要。[5]最广泛的致突变活性生物测定方法是AMES试验。该测试使用鼠伤寒沙门氏菌细菌,该细菌具有预先存在的突变,可阻止组氨酸的合成。没有组氨酸,细菌就无法生长。当一种化学物质显示出致突变活性时,DNA反应性就很明显,这会引起突变,使细菌回复到可以产生组氨酸的状态,从而开始生长。[6]因此,细菌生长的程度为受试化学品的致突变潜力提供了定量和定性的指示。使用计算毒理学预测Ames测试结果是可能的,但是,许多当前使用的方法通常依赖于化学系统的专家知识。[7]此类计算机模拟系统,例如Derek Nexus,基于所谓的“结构警戒”将化学品分类(例如,DNA反应性,参见图1)。[8,9]
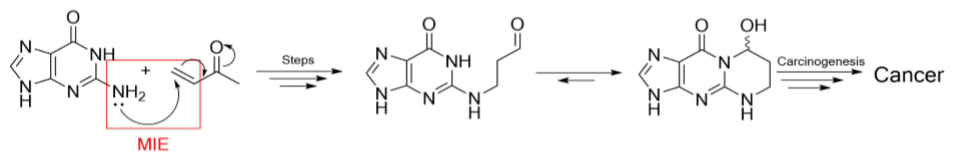
Figure 1. 1,4-Michael受体与DNA核碱基鸟嘌呤的反应。红色框表示分子引发事件——导致不良毒理学反应的一系列反应的第一步。在这种情况下,不良结果是致癌作用。
DNA反应性类别内结构警戒的实例仅列举了α,β-不饱和羰基、酰卤和异氰酸酯。然而,基于官能团存在与否的分类方法(或“结构警戒”)并不能充分解释与致突变性相关的基本化学机理。外源性化学品与DNA之间的初始反应可描述为“分子引发事件”。[10]此反应可触发“不良结果途径”(AOP),其定义为初始暴露与不良结果之间的一系列事件。例如癌症,呼吸衰竭等。[9]透彻了解分子引发事件背后的基本化学原理,将使我们能够更好地预测毒理学实验方法(例如AMES试验)的结果。这有助于实现对候选药物进行高通量的计算机筛选,从而使预测更快、更便宜。量子化学提供了探测分子引发事件的必要工具。特别是,密度泛函理论(DFT)过渡态建模已被证明可能够全面评估化学反应机理的空间和能量细节。[11]这通常通过计算活化能来评估哪个反应路径可能进行来实现。Goodman等人先前已经证明,DFT的活化能垒可用于预测与DNA形成共价键的多种Michael受体的AMES测试结果。[12]本研究对Goodman等人发表的模型进行了改进,并证明最低未占据分子轨道(LUMO)能量对于评估Michael受体型化合物的致突变性潜力具有显著的预测性。
材料与方法
30个α,β-不饱和羰基化合物的AMES测试结果取自OECD QSAR Toolbox以及Perez-Garrido及其同事发布的数据集。[13,14]其中19个化合物的数据是由Goodman等人在进行本研究时获得的。[12]其中16个化合物中归类为AMES阴性,14个化合物归类为AMES阳性。所有实验AMES测试数据均未使用S9酶激活系统。密度泛函理论(DFT)用于研究Michael受体数据集与模型亲核试剂之间的反应(见Figure 2)。
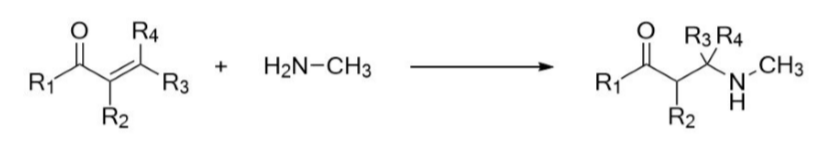
Figure 2. 1,4-Michael受体与甲胺的通式反应
首先计算“分子引发事件”的活化能。为了确保计算时间的合理,选择了甲胺作为亲核试剂来模拟含氮的DNA碱基(见图3)。化合物16和23分别截取自柠檬醛和白头翁(pulegone)。[15]在明显构象柔性的情况下,使用Schrödinger的Macromodel(ver 11.3)和MMFF力场进行构象搜索。[16-18]反应物和过渡态构象均使用Macromodel的low mode采样方法[19]获得。然后反应物和过渡态用Gaussian 16(Reversion A.03)的DFT方法在B3LYP/DEF2-TZVPP水平下(考虑Grimme的D3色散条件下)进行优化。[20-22]在整个工作过程中,使用了IEF-PCM考虑了水的溶剂效应。IEF-PCM模型先前已广泛用于有机化学反应的建模。[23,24]还用GoodVibes法[26]计算了温度和浓度校正的准谐波自由能(Grimme近似[25]),其中包括振动比例因子1,温度310.15 K和浓度1 mol DM-3。[26]化合物的最低能量基态和过渡态的电子补充信息用ESIGen生成。[27]
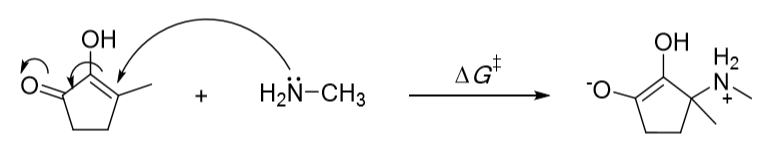
Figure 3. 2-Hydroxy-3-methylcyclopent-2-enone与甲胺的反应
结果与讨论
活化能
Table 1.数据汇总
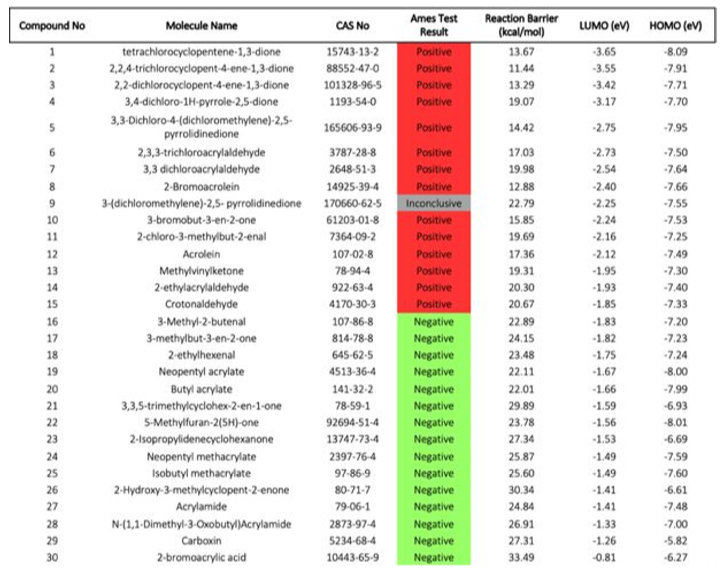
总共获得了30个化合物的活化能(见Table 1)。结果表明,活化能范围很广,从11.4至33.5 kcal/mol(见Figure 4)。AMES阳性化合物的范围为11.4-20.7kcal/mol。 AMES阴性化合物的范围为22.0-33.5 kcal/mol,两个离群值分别为13.7和22.8 kcal/mol(请参见下面的讨论)。 AMES阳性化合物的平均活化自由能为16.8 kcal/mol,而MES阴性化合物的平均活化能为24.9 kcal/mol。AMES阳性和AMES阴性化合物的平均活化能之间存在8.1 kcal/mol的能隙。这将Goodman等人发布的19个化合物的最高AMES阳性化合物和最低AMES负化合物的活化自由能隙从2.2 kcal/mol提高到2.9 kcal/mol。这些结果是使用色散校正的B3LYP泛函和更大的基组来实现的。[12,28]在模型扩展后,最高活化能(22.8 kcal/mol)的AMES阳性化合物是化合物9,该化合物的能垒大小位于AMES阴性化合物的预期范围内。然而,Haddon等人先前的研究表明,化合物9确实对鼠伤寒沙门氏菌TA100菌株呈AMES阳性。尽管分类如此,但化合物9表现出极低的摩尔致突变性(0.24 rev/nmol),这解释了为什么其活化能位于AMES阴性区域内。除此之外,在同一研究中,AMES阴性、非致突变化合物被引用为具有相似的摩尔致突变性(小于0.3 rev/nmol)。因此,将化合物9从模型中删除,作者建议完全重新评估化合物9的AMES测试。另一个异常值是化合物1,最初是从OECD QSAR Toolbox中获得被认为是AMES阴性化合物,其活化能为13.7kcal/mol。在检查化合物的活化能和LUMO能量后,很明显地化合物1被预测为AMES阳性化合物。为了探索该结果,研究了化合物1的现有文献。发现一项研究显示在AMES试验中呈阳性活性,从而验证了我们模型的预测结果。[29]该化合物的AMES试验结果也相应地改变了。
本研究中的化合物包括一种羧酸,即化合物30。羧酸的标准酸度为2-5 pKa,在生理pH值下,化合物30的主要形式将是离子化的羧酸。[30] 因此,对化合物30的共轭碱进行了计算。该化合物的AMES实验测试结果为阴性,与活化能(33.5 kcal/mol)显示出良好的一致性。
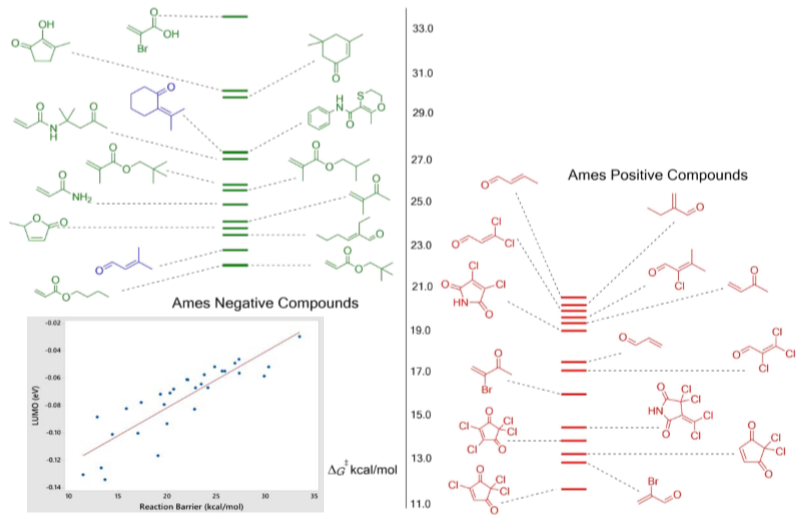
Figure 4. Michael受体与甲胺的反应活化能排序
EHOMO与ELUMO
本研究计算了30个化合物的HOMO与LUMO能量。尽管文献上已有成功的尝试,但HOMO能量并未显示出作为致突变性毒理学描述符的预测潜力(参见表1)。 [31,32]然而,Michael受体的LUMO能量被证明是一个非常有前途的描述符。LUMO能量的范围从-3.65 eV(计算的最低LUMO能量)到-0.81 eV(计算的最高LUMO能量)(请参见表1)。AMES阳性化合物的LUMO能量在-3.65 eV至-1.85 eV范围之间,AMES阳性化合物的平均LUMO能量为-2.57 eV。 AMES阴性化合物的范围为1.83 eV至-0.81 eV,AMES阴性化合物的平均LUMO为-1.50 eV。显然,与AMES阴性化合物相比,AMES阳性化合物的LUMO能量更低。我们的研究结果表明,作为一种毒理学指标,LUMO能量对于准确预测Michael受体的AMES测试结果显示出巨大的潜力。除此之外,LUMO能量可以从量子化学计算中轻松地获得。[33]此外,LUMO能量与计算出的活化能具有统计学意义的显著相关性(请参阅补充资料)。回归分析得出r2 = 0.75,表明活化能和LUMO能量之间具有良好的相关性。同样,相关性系数“ r”等于0.89,表明随着反应势垒的增加,LUMO能量也同时增加。
与现有方法的比较
为了评估模型的性能,使用预存在(Q)SAR软件来预测数据集中化合物的体外AMES测试结果。使用“Toxicity Estimation Software Tool”(TEST)中的“Nearest neighbor”方法预测AMES测试结果。[34]这种方法将待测试化学品与QSAR训练集中的三种最相似的化合物进行比较以预测致突变性。TEST可以正确预测14种AMES阳性化合物中的9种,以及15种AMES阴性化合物中的11种。尽管TEST表现出相对较好的性能,但我们的模型预测AMES具有更高的一致性。此外,还更深入了解毒物-靶标相互作用相关的基础化学原理。总体而言,LUMO能量和活化能都有望用作主要(Q)SAR指标,并且很容易满足毒理学政策中的指导原则。例如,国际协调委员会(ICH)宣布应使用两种互补的计算机模拟方法来评估药品中杂质的致突变性[35]。
结论
计算了30个迈克尔受体的活化自由能和HOMO/LUMO能量。AMES阳性化合物活化能范围为11.4-20.7 kcal/mol,而AMES阴性化合物的活化能范围为22.0-33.5 kcal/mol。 AMES阳性化合物的LUMO能量范围为-3.65至-1.85 eV,AMES阴性化合物的LUMO能量范围为-1.83至0.81eV。化合物1最初被归类为AMES阴性,但是在用我们的模型预测和文献评估后,已将其更正为AMES阳性。我们可以有把握地预测,ELUMO小于-1.85 eV和活化能小于20.7 kcal/mol的Michael受体将是AMES阳性化合物,而活化能大于22.0 kcal/mol和ELUMO大于-1.83 eV的Michael受体将是AMES阴性化合物。
这些结果表明,密度泛函理论过渡态模型对于理解分子引发事件以及致突变性的内在化学起源具有很大的潜力。当将活化能和LUMO能量组合为描述符时,它们对Michael受体型化合物中的AMES试验分类显示出优异的预测潜力。考虑到上述信息,作者建议在研究预先存在的和新合成的1,4-Michael受体的致突变潜力时,应同时彻底地研究活化能和LUMO能量。将来,该框架可应用于已知与DNA反应的不同组的化合物,例如希夫碱试剂。然而,不同的化合物组别之间可能有不同的活化能垒范围与LUMO能量范围。因此,DFT过渡状态建模应该独立地应用于每个化学类别。总之,我们开发了一种快速、低成本的框架, 在评估药学上感兴趣的Michael受体的致突变性风险上比现有方法更优。活化能和LUMO能量对Michael受体中的致突变风险显示出显著的预测性,因此应该将这些描述符纳入到现有或未来的毒理学QSAR模型中。
相关主题(请点击标题看原文)
- Gaussian教程 | 搜索过渡态
- Gaussian教程 | 绘制分子轨道
- 选择性JAK3抑制剂PF-06651600的设计
本文介绍了如何用Gaussian进行过渡态搜索,计算活化能。在教程的指导下,应该可以重现上述的活化能计算。
本文描述了波函数、原子轨道、分子轨道与前线轨道的基本概念,以及如何用Gaussian与GaussView计算、绘制、可视化、观察分子轨道。自然少不了介绍如何计算前线轨道能量EHOMO与ELUMO。在教程的指导下,应该可以重现上述的HOMO与LUMO能量计算。
本文介绍了辉瑞公司选择性JAK3抑制剂PF-06651600的设计过程。PF-06651600是一种Michael受体化合物与JAK3的CYS909巯基发生共价作用。本文为共价药物设计提供了一般性的参考。如果配合上本文的AMES预测,可以帮助建立AMES化合物的警戒系统。


