
摘要:本文描述了波函数、原子轨道、分子轨道与前线轨道的基本概念,以及如何用Gaussian与GaussView计算、绘制、可视化、观察分子轨道。
作者:陈宇
时间:2017-11-27
一.计算分子轨道(molecular orbitals)的基本原理
1. 基本概念
波函数(wave function):在量子力学中,粒子的状态用波函数(满足特定条件的函数)来描述,波函数本身没有明确的物理意义,但波函数的平方描述了粒子在特定区域出现的概率。波函数能够通过求解薛定谔方程得到,理论上,当确定了一个研究对象的波函数后,就能够获得研究对象的所有性质。
原子轨道(atomic orbitals):原子轨道是指原子中电子的所有可能运动状态,对于单电子原子体系(也就是氢原子),我们能够精确求解薛定谔方程得到一系列正交化的波函数(也就是原子轨道)。在杂化轨道理论中,原子之间的成键过程被理解为在一定规则下原子轨道的有效重叠,而形成的分子中,电子是被定域在原子周围的。
分子轨道(molecular orbitals):分子轨道是指分子中电子的所有可能运动状态,在分子轨道理论中,分子中的电子被设想为离域在整个分子体系中。分子轨道波函数通常被表示为组成分子的所有原子的原子轨道的线性组合,能够通过近似求解薛定谔方程得到。
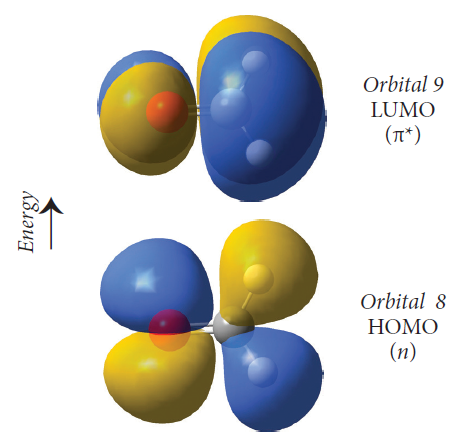
前线轨道(frontier orbitals):前线轨道理论认为,在一个分子的所有分子轨道中,能量最高的占据轨道(HOMO)和能量最低的非占据轨道(LUMO)对分子的反应和性质起着决定性的作用(图1),这些轨道也被统称为前线轨道(也包括SOMO轨道,指的是单电子占据轨道)。对大多数化学反应而言,在满足分子轨道对称性的条件下,反应在一个反应物的HOMO与另一反应物的LUMO能够产生最大重叠位置及方向上发生。
图1. 分子的HOMO和LUMO轨道
2. 生成分子轨道的方法
以上的理论表明:对分子轨道具体信息(包括分子轨道能量和分子轨道形状)的了解有助于我们对分子反应性和其他分子性质的了解, 而其中尤其重要的就是分子的HOMO和LUMO轨道。
为了生成分子轨道,我们需要首先求解分子体系的薛定谔方程。实际上,求解薛定谔方程几乎是任何计算任务的先决步骤。但通常情况下,输出文件并不会给出分子轨道的具体成分和相应的系数。Gaussian程序能够通过调用Pop关键字打印出有关分子轨道的具体信息,并能够通过GaussView可视化这些信息(也就是绘制分子轨道图)。
3. 绘制分子轨道图的几个步骤
绘制分子轨道图通常需要进行如下三个步骤:
- 利用opt关键字优化得到分子的稳定构型
- 用优化得到的结构作为初始结构,计算单点能量,并打印出分子轨道数据
- 利用GaussView绘制分子轨道图
二.利用Gaussian程序绘制分子轨道的操作步骤
下面我们将以NH3分子(氨气分子)为例,介绍如何绘制HOMO和LUMO轨道。
第一步:利用opt关键字优化NH3分子的稳定构型。
利用opt关键字优化NH3分子的输入文件:
1 2 3 4 5 6 7 8 9 10 11 12 | %nprocshared=8 %mem=1GB # b3lyp/6-31g(d) opt freq opt 0 1 N 2.14032153 -0.25625990 -1.57584313 H 1.99433846 -1.18548792 -1.91529008 H 3.09370723 0.00673111 -1.72380070 H 1.54024195 0.37583019 -2.06611512 [注意:此行要用空白行替换,之前所有空白行不可省略] |
图2. NH3的优化与频率计算输入文件
第二步:用优化得到的结构作为初始结构,计算单点能量,并打印出分子轨道数据。
输入文件如下:
1 2 3 4 5 6 7 8 9 10 11 12 13 | %nprocshared=8 %chk=NH3-mo.chk %mem=1GB # hf/6-31g(d) pop=Regular gfinput MO 0 1 N 0.00000000 0.00000000 0.11934900 H 0.00000000 0.93858100 -0.27848200 H -0.81283500 -0.46929100 -0.27848200 H 0.81283500 -0.46929100 -0.27848200 [注意:此行要用空白行替换,之前所有空白行不可省略] |
图3. MO计算输入文件
其中第4行的关键字gfinput指示程序输出所使用基函数的具体参数。
第4行的关键字pop=Regular则指示程序输出能量最高的五个占据分子轨道和能量最低的五个非占据分子轨道的具体参数(如果想输出所有分子轨道参数可使用关键字pop=full),这些数据是我们研究分子轨道所需要的,在本例中的输出如下:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 | Orbital symmetries: Occupied (A) (A) (E) (E) (A) Virtual (A) (E) (E) (E) (E) (A) (A) (E) (E) (A) (E) (E) (A) (E) (E) (A) The electronic state is 1-A. Alpha occ. eigenvalues -- -15.54186 -1.13458 -0.61899 -0.61899 -0.42388 Alpha virt. eigenvalues -- 0.21866 0.32002 0.32002 0.89589 0.89589 Alpha virt. eigenvalues -- 0.97010 1.20593 1.21539 1.21539 1.38023 Alpha virt. eigenvalues -- 1.96609 1.96609 2.24843 2.74753 2.74753 Alpha virt. eigenvalues -- 4.08187 Molecular Orbital Coefficients: 1 2 3 4 5 O O O O O Eigenvalues -- -15.54186 -1.13458 -0.61899 -0.61899 -0.42388 1 1 N 1S 0.99500 -0.19706 0.00000 0.00000 -0.06632 2 2S 0.02469 0.40932 0.00000 0.00000 0.15611 3 2PX 0.00000 0.00000 0.05707 0.46663 0.00000 4 2PY 0.00000 0.00000 0.46663 -0.05707 0.00000 5 2PZ -0.00158 -0.09058 0.00000 0.00000 0.55067 6 3S -0.00166 0.43454 0.00000 0.00000 0.28323 7 3PX 0.00000 0.00000 0.03405 0.27841 0.00000 8 3PY 0.00000 0.00000 0.27841 -0.03405 0.00000 9 3PZ 0.00095 -0.05132 0.00000 0.00000 0.48389 10 4XX -0.00348 0.01404 -0.02099 0.00257 0.00828 11 4YY -0.00348 0.01404 0.02099 -0.00257 0.00828 12 4ZZ -0.00328 0.00118 0.00000 0.00000 -0.03307 13 4XY 0.00000 0.00000 -0.00297 -0.02424 0.00000 14 4XZ 0.00000 0.00000 -0.00497 -0.04066 0.00000 15 4YZ 0.00000 0.00000 -0.04066 0.00497 0.00000 16 2 H 1S 0.00023 0.13547 0.27290 -0.03338 -0.06814 17 2S 0.00058 0.01012 0.16944 -0.02072 -0.05220 18 3 H 1S 0.00023 0.13547 -0.16536 -0.21965 -0.06814 19 2S 0.00058 0.01012 -0.10267 -0.13638 -0.05220 20 4 H 1S 0.00023 0.13547 -0.10754 0.25303 -0.06814 21 2S 0.00058 0.01012 -0.06677 0.15710 -0.05220 6 7 8 9 10 V V V V V Eigenvalues -- 0.21866 0.32002 0.32002 0.89589 0.89589 1 1 N 1S -0.12464 0.00000 0.00000 0.00000 0.00000 2 2S 0.10067 0.00000 0.00000 0.00000 0.00000 3 2PX 0.00000 -0.31118 -0.12768 -0.34989 -0.06873 4 2PY 0.00000 0.12768 -0.31118 0.06873 -0.34989 5 2PZ -0.15485 0.00000 0.00000 0.00000 0.00000 6 3S 1.95248 0.00000 0.00000 0.00000 0.00000 7 3PX 0.00000 -1.02365 -0.42002 1.13716 0.22337 8 3PY 0.00000 0.42002 -1.02365 -0.22337 1.13716 9 3PZ -0.51646 0.00000 0.00000 0.00000 0.00000 10 4XX -0.03973 0.00083 -0.00203 -0.02309 0.11756 11 4YY -0.03973 -0.00083 0.00203 0.02309 -0.11756 12 4ZZ -0.03771 0.00000 0.00000 0.00000 0.00000 13 4XY 0.00000 -0.00235 -0.00096 0.13575 0.02667 14 4XZ 0.00000 0.01549 0.00636 0.08733 0.01715 15 4YZ 0.00000 -0.00636 0.01549 -0.01715 0.08733 16 2 H 1S -0.02902 -0.01920 0.04678 0.15017 -0.76450 17 2S -0.96877 -0.66395 1.61816 -0.01960 0.09979 18 3 H 1S -0.02902 -0.03092 -0.04002 0.58699 0.51230 19 2S -0.96877 -1.06939 -1.38408 -0.07662 -0.06687 20 4 H 1S -0.02902 0.05011 -0.00677 -0.73716 0.25220 21 2S -0.96877 1.73335 -0.23408 0.09622 -0.03292 |
图4.分子轨道计算输出
其中,第二行,连同中间行的数字1,2,3,4,5,6,7,8,9,10分别表示能量由低到高排列的分子轨道,数字1,2,3,4,5下面对应的字母O表示这些轨道被电子所占据;而数字6,7,8,9,10下面的V则表示这些轨道是未被电子占据的空轨道;Eigenvalues(本征值)所在行的数值是这些分子轨道的能量;能量下面的所有行则表示组成分子轨道的原子轨道的系数,这些数值为我们绘制分子轨道提供了基本的数据。
需要注意的是,在这里我们需要保存chk文件(也就是%chk=NH3-mo.chk)以便绘制分子轨道图。(还需要注意的是如果使用Linux版的Gaussian,在计算完成之后需要在Linux命令行输入 formchk NH3-mo.chk 将chk文件转化为fchk文件。如果使用的是Windows版本的Gaussian则可以直接使用chk文件)。
第三步:利用GaussView绘制分子轨道图。(在这里我们使用的Linux版本的Gaussian程序)
1. 分子轨道编辑对话框的使用
双击生成的NH3-mo.fchk文件(对于Windows版双击chk文件)。点击箭头指示的位置,调出分子轨道编辑对话框。
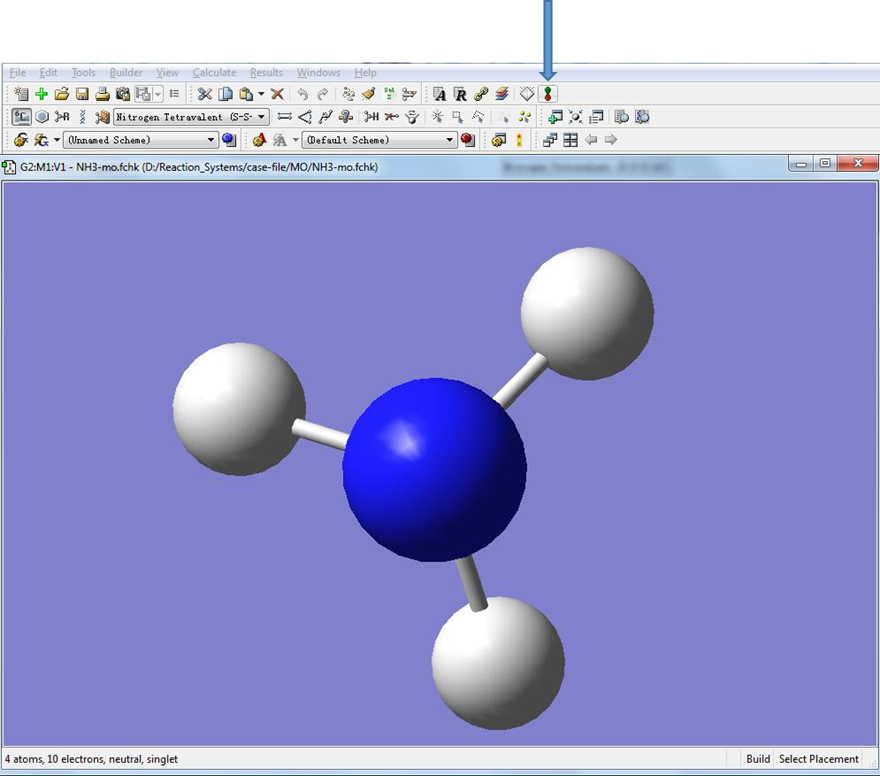
图5. 分子轨道编辑器
2. 分子轨道的可视化
点击箭头所指的区域,进入Visualize分子轨道对话框,并选中5和6所指示的分子轨道
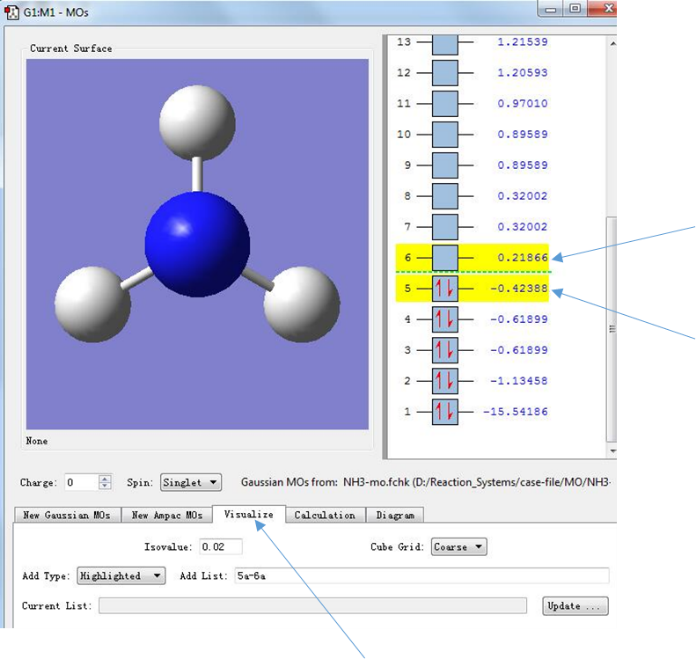
图6.分子轨道的选择
在这里需要说明的是图中右侧显示的便是按能量由低到高(从下往上)排列的分子轨道,以及其中占据的电子数和相应的轨道能量(后面会有详细的讨论)。 值得一提的是分子的电子能并不等于所有占据轨道的能量之和,因为分子轨道能量并未考虑到电子之间的排斥作用。
3. 分子轨道的绘制
在这里我们将等值面Isovalue值设定为0.07(默认为0.02,通常情况下,Isovalue值以能够突出分子轨道特征为准),点击Update,即可生成分子轨道图(图8)。下面我们将说明能够从图中得到的一些重要信息:
(1)首先需要澄清的一点是,显示的分子轨道并不是“电子云”,电子云描述的是电子出现的概率,而分子轨道则是指电子运动的波函数,对波函数取平方才能得到电子在某处出现的概率。
(2)可以看到分子轨道出现红绿两种颜色,实际上这代表了波函数不同的相位(在波函数中体现为正负号)。由于电子密度与波函数的平方相关,因此,波函数相位的改变并不影响电子密度的分布。但相位却决定了原子间形成成键轨道还是反键轨道,下面以两个氢原子形成氢分子为例说明相位如何影响轨道的形成(图7)。
H原子的一个电子占有1s(球形)轨道,当两个H原子相互靠近时,同相位波函数(均为正号,也可以均为符号)重叠使得两个原子之间的电子密度增大,从而得到一个能量比重叠前的原子轨道能量都低的分子轨道(也称为成键轨道),两个电子将填充在成键轨道中,从而形成一个稳定的氢分子;而当两个H原子相互靠近时,异相位波函数(一个正号,一个负号)重叠使得两个原子之间的电子密度减小,从而得到一个能量比重叠前的原子轨道能量都高的分子轨道(也称为反键轨道)。
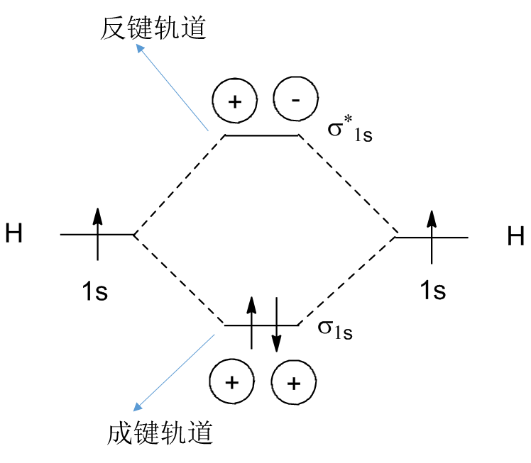
图7. 成键轨道与反键轨道
(3)依据电子分布的位置,我们也可以判断电子所处的轨道类型,图8中,序号5后面箭头指示的红色小框表明,此时显示的分子轨道是能量为-0.42388 hartree的轨道,而代表这条轨道的小方框中两条指向相反的箭头则表示在这条轨道中填充了两个自旋相反的电子(小方框中若没有红色箭头,表明这条轨道中没有填充电子),可以看出这条轨道是填充了电子的能量最高的轨道,也就是HOMO轨道。从图中来看,电子分布集中在NH3分子的C3v对称轴上(集中在N原子周围),而没有处于N-H连线上(这说明HOMO轨道的电子并没有参与N-H的成键),因此我们认为HOMO轨道被一对孤对电子所占据(孤对电子是指原子的外电子层中未成键的电子对)。
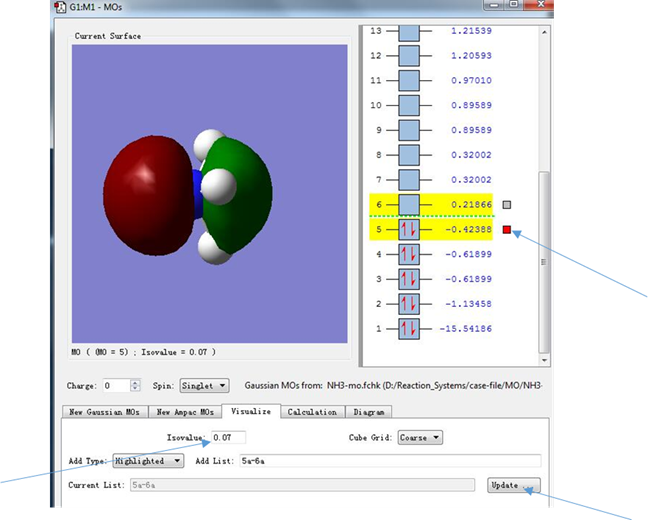
图8. HOMO/LUMO分子轨道的可视化
(4)接下来我们点击序号为6的分子轨道最右侧的箭头指示的小灰框,使其变成红色,就可以显示序号为6的分子轨道。此时显示的分子轨道能量为0.21866 hartree,而代表这条轨道的小方框中并没有红色箭头,表明这条轨道中没有填充电子,可以看出这条轨道是没有填充电子的能量最低的轨道,也就是LUMO轨道。在LUMO轨道中,电子密度主要集中在N-H原子的连线上,但又没有处于N-H原子之间,这表明LUMO轨道处于N-H反键位置。
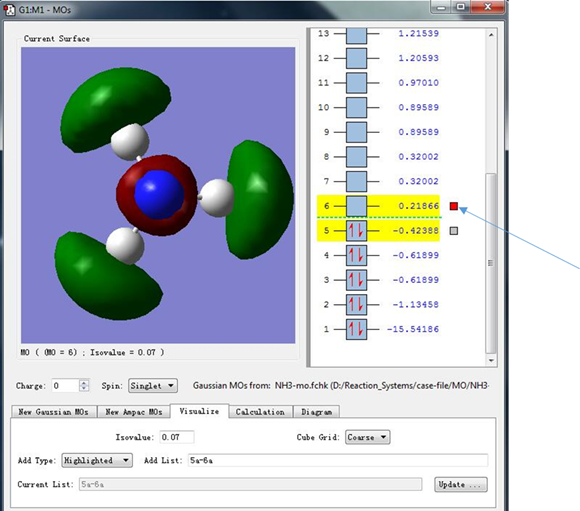
图9. 分子轨道能量与电子填充
(5)有时我们可能也会关心除HOMO和LUMO轨道外的其他分子轨道,为了绘制其他的分子轨道,我们可以选中感兴趣的分子轨道(会显示黄色标志,如下图选中序号为3,4,5,6,7的轨道),然后点击Update,即可绘制选中的分子轨道图,下图中箭头指示的分子轨道是序号为4的分子轨道,可以看出这个轨道被两个电子所占据,若按照能量由高到低排列,这个轨道的能量在所有电子占据轨道中排列第二(这类分子轨道也称为HOMO-1轨道)。在HOMO-1轨道中,电子密度主要集中在N-H原子的连线上,并且处于N-H原子之间,这也表明HOMO-1轨道处于N-H成键位置。
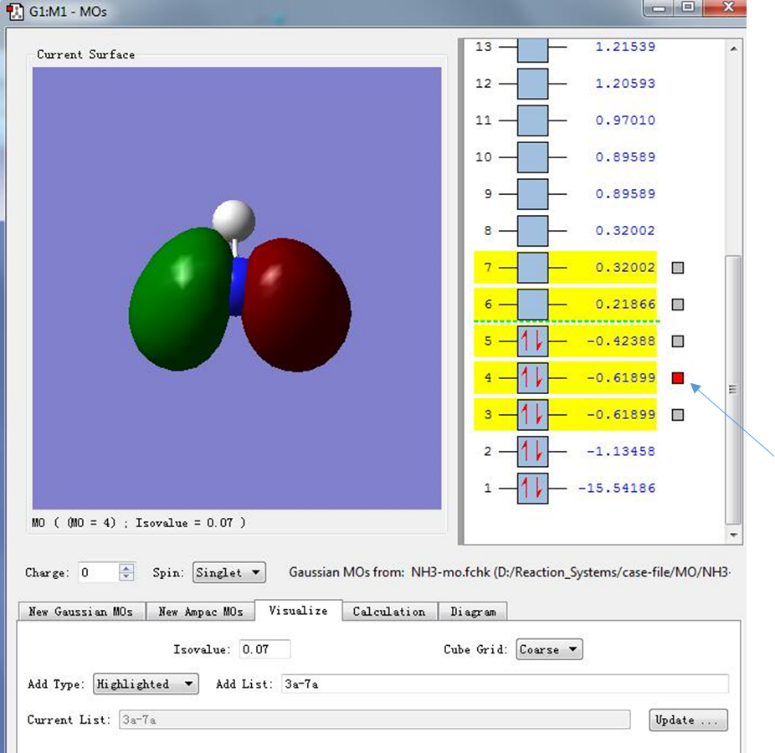
图10. 分子轨道的可视化
相关主题
- Gaussian教程:http://blog.molcalx.com.cn/tag/gaussian
参考材料
- http://gaussian.com/population
- 《基础量子化学与应用》 刘靖疆
- https://en.wikipedia.org/wiki/Molecular_orbital
- 《化学反应与电子轨道》福井谦一


