
摘要:本文作者开发了一个鲁棒的系统DP4-AI,用于自动处理和归属原始13C和1H-NMR数据,已集成到计算有机分子结构确证工作流中。从具有未定义的立体化学或其它结构不确定性的分子结构开始,该系统允许完全自动化的结构确证。开发了使用目标模型选择的NMR峰捡拾方法和用于将计算的13C和1H-NMR化学位移与NOISY实验NMR数据峰匹配的算法。使用具有挑战性的分子进行严格评估结果表明:DP4-AI的处理速度提高了60倍,节省了科研人员大量的时间。 DP4-AI代表了NMR结构确证方法的飞跃也是DP4功能的飞跃。使得数据库和大量分子的高通量分析成为可能,而在这以前是不可能进行的,并为通过机器学习发现新的结构信息铺平了道路。已经为新功能配上了直观的GUI,软件是开源的,获取方法:https://github.com/KristapsE/DP4-AI。
编译:肖高铿/2020/09/13
原文:Howarth, A.; Ermanis, K.; Goodman, J. M. DP4-AI Automated NMR Data Analysis: Straight from Spectrometer to Structure. Chem. Sci. 2020, 11 (17), 4351–4359. https://doi.org/10.1039/D0SC00442A.
前言
在合成有机化学和天然产物化学中,分子结构确证是一个具有挑战性的问题。结构相近的异构体(例如区域异构体和保护基定位)和非对映异构体通常在它们的1D-NMR光谱中仅表现出微小的差异,使得结构确证和相对立体化学确证非常困难。这可以通过诸如NOESY谱或合成天然产物的异构体并将所得观察到的NMR谱与公开的那些进行比较来解决。这两种方法都非常昂贵且耗时。
一个有吸引力并且现在已经建立的备选方案1,2是计算NMR预测。该方法使用DFT计算未确定结构所有非对映异构体的NMR化学位移,将预测结果与公开发表的光谱进行比较,并使用诸如相关系数、平均绝对误差(MAE)、校正平均绝对误差(CMAE)等参数进行评估3。DP4分析特别有效,因为它不仅预测分子的相对立体化学和其他变化。 而且也使用贝叶斯定理给出了每个候选分子是正确分子的概率(假设所提供或生成的结构之一是正确的)3,4。DP4已经成功地用于确证许多天然产物类似分子分子、合成中间体、天然产物片段以及药物的立体化学5-10,并进一步在DP4+和J-DP4分析中探索11,12。
自从发表以来,DP4计算已经得到简化,并且用户输入也得到最小化,因为现在所有计算都由Python程序PyDP 4自动管理11-13,只有那些立体化学不明确和归属实验1D 13C和1H NMR光谱的分子结构才需要用户人工输入。在使用该程序进行相对立体化学确证时,用户工作量最大的部分是NMR谱的归属。这不仅费时费力,且易于出错14。NMR光谱的自动解释是多年来分析化学的主要目标15。这方面大量工作集中在开发CASE(计算机辅助结构确证)软件16-18用于自动2D结构确证而不是对已知结构进行自动归属原子这个重复劳动。典型地,结构确证不仅需要提供1D NMR光谱,而且还要提供多个2D NMR光谱19。
少数商业软件包提供专家指导的1H-NMR谱的NMR归属算法,比如著名的软件Mestrelab Mnova20。该软件主要用于辅助用户解释NMR谱,但不能对原始NMR数据自动处理与归属。
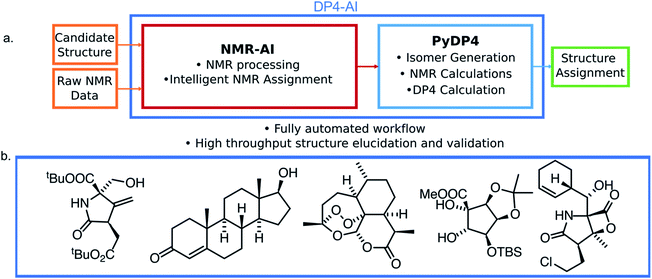
Figure 1. (a)DP4-AI的结构。本系统提供了一种全自动立体化学结构确证方法,用户仅需要将原始的NMR数据作为输入即可。(b)几个用DP4-AI全自动进行立体化学预测的算例。
本文呈现了一种全自动稳健处理、归属1H和13C-NMR光谱的系统(图1)。该程序的示意图如图2所示。它用1D 1H和13C-NMR光谱进行自动化相对立体化学和结构模糊预测。化学位移值使用DFT GIAO方法计算。化学位移还可以使用诸如NWChem21和Tinker22这样的开源免费软件来进行计算。
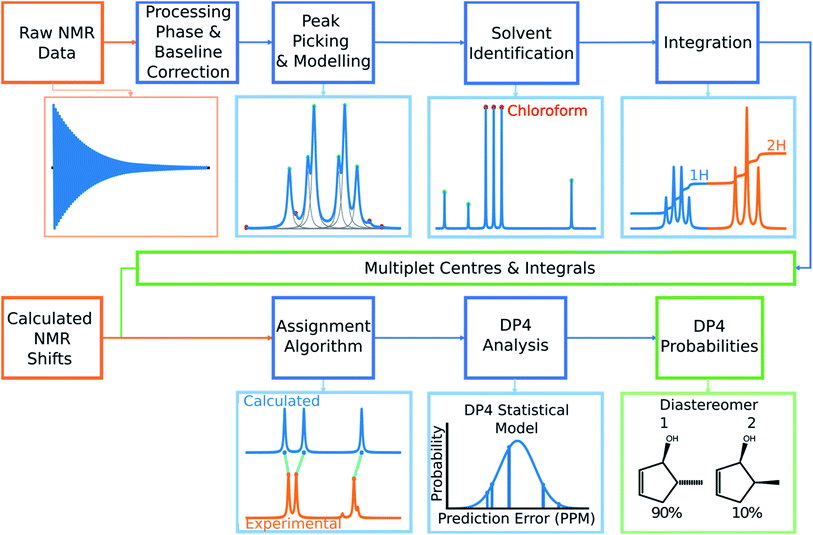
Figure 2. DP4-AI的结构。原始NMR数据经过一系列处理得到实验化学位移值与积分。程序然后用DFT计算每个原子的化学位移并将之归属到实验的峰位上。归属之后计算每个差向异构体的DP4概率
自动化的DP4-AI令人兴奋,因为这使得以前不可能的高通量数据库和大量分子分析成为可能。 此外,NMR谱的自动处理和归属减少了合成的时间限制,将更多的机会用于化学发现。 此外,该系统在不久的将来为开发更复杂的NMR实验自动解释的开发提供框架,并且可以与CASE软件一起使用,以解决分析数据中的结构确证问题。
计算方法
DP4计算沿用了之前的方法3,11–13。分子力学计算采用MacroModel(Version 9.9),构象搜索采用Low Mode与Monte Carlo搜索混合算法采用MMFF力场在气相中进行29,30。设置MacroModel的步数,以便所有低能量构象体至少被发现5次。量子力学计算采用Gaussian 09软件。GIAO方法用于NMR屏蔽常数的计算31-33。NMR化学位移采用mPW1PW91泛函以及6-311G(d)基组进行计算,这是因为在DP4计算时优化得到的12。对于含碘的分子,使用def2-SVP基组36,37。所有的DFT计算都使用隐式PCM溶剂模型38。分子的几何结构也在B3LYP/6-31G(d)理论水平上优化,然后在M06-2X/def2-TZVP理论水平计算单点能。
计算用Python 3.7编写的PyDP4 Python脚本管理,该脚本现在是DP4-AI的一部分。DP4-AI可从http://www-jmg.ch.cam.ac.uk/tools/nmr/和GitHub https://github.com/KristapsE/DP4-AI/获得。NMR处理的某些部件使用了NMRglue软件包41。
程序描述
自动化的NMR处理让用户无需费力地编写NMR描述,从根本上提高了该过程的生产效率。为了将分子中的原子归属为NMR光谱中的峰,必须从原始NMR数据中提取峰的位置和积分值,如图2所示。由于所有NMR图谱都不相同,并且每个阶段的处理会对后续处理产生影响,所以全自动NMR数据的处理和分析非常复杂。尽管存在这些挑战,但DP4-AI仍被设计为尽可能强大且可靠地处理NMR数据。程序的此部分概述如下,而更详细的描述参见ESI(第S2.1节)。
在执行傅立叶变换之后,谱图可能会显示相位误差,必须在进一步处理之前对其进行校正。不幸的是,没有一种现有方法能够按要求可靠地对光谱测试集进行相位调整。为了解决这个问题,Wang等人42开发了一种信号混合方法,将相变算法ACME43的基于熵的目标函数与Zorin等人44开发的鲁棒的加权线性回归方法(WLR)结合在一起,以解决这一问题。
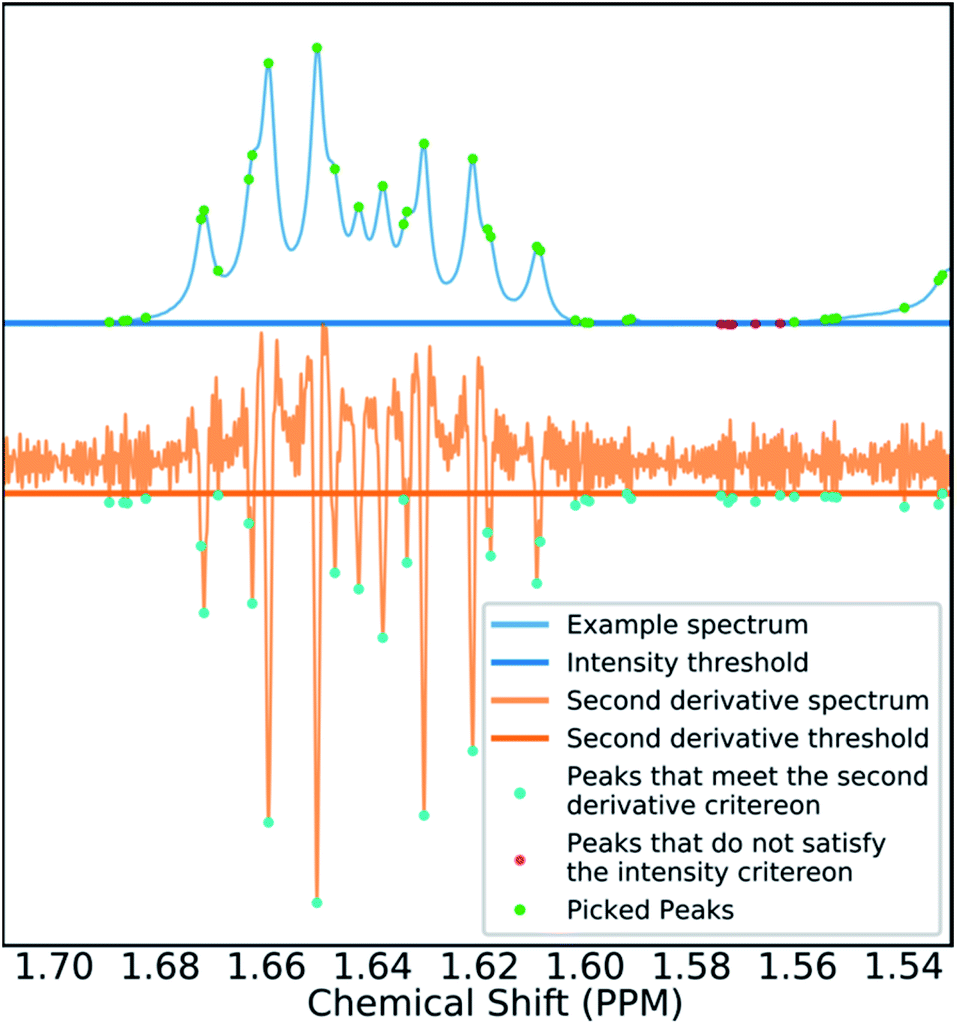
Figure 3. 梯度峰捡拾过程:如果一个峰的二阶导(黄色)低于一定阈值并且高于强度阈值(蓝色)则捡拾该峰。最终被捡拾出的峰用绿色高亮显示
许多光谱还发现有基线扭曲,务必进行消除处理。 Wang et al.开发42的算法的修改版本已被合并到最终的程序中(ESI第S2.1.4†节)。对于1H谱,首先使用光谱一阶和二阶导进行峰的捡拾。若一阶导为零而第二阶导中为极小值则认为发现潜在的峰。如果一候选峰在幅度阈值之上并且其二阶导在阈值之下,则捡拾该峰。 这些阈值是自适应的,因为它们被设置为噪声标准偏差值的数倍。这种峰捡拾方法可以将两个阈值都设置得非常低,从而尽可能多地滤除噪声,同时尽可能少地丢失信号。 此外,使用导数可确保基线独立性。 这个过程总结在图3中。
在1H谱中,必须将信号峰分组在一起以建立多重峰中心的位置。预计在1H谱中的质子之间可以看到的最大耦合常数约为18Hz。间隔小于18 Hz的任何峰都可以组合成多重峰。为了避免遗漏任何信号峰,特意将峰捡拾阈值信噪比的阈值选取阈值设置得非常低。但是,这同时增加了噪声峰值被误认为信号峰值的可能性,并且可能导致过多峰分组(ESI第S2.1.6节)。
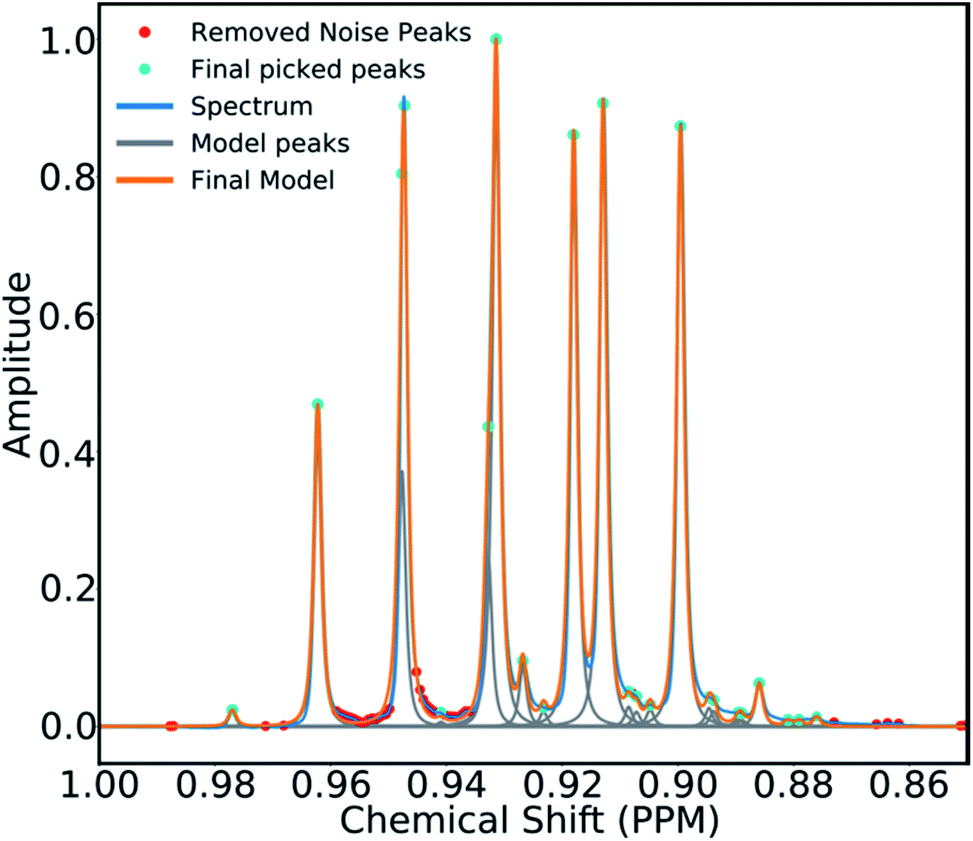
Figure 4. 多重峰(蓝色)和反卷积模型(橙色)示例。信号峰用青色突出显示,确定为噪声的峰用红色突出显示。
为了减轻这个问题,开发了一种利用目标模型选择来消除噪声的算法。间隔小于18Hz的捡拾峰被分组在一起以定义信号覆盖的区域。对于每个区域,多重广义洛伦兹线形函数用来构建线形模型45。每个区域的模型中的参数会反复变化,直到模型的积分收敛到光谱对应区域的1%以内。为避免过拟合,然后测试描述每个峰的参数组的信息含量。构造一个新模型,而没有依次具有每个线形函数。如果模型的贝叶斯信息准则(ESI第S2.1.6†节)降低了一个阈值以上,则认为这些参数描述了噪音峰(因为它们不会增加模型的信息内容)并被删除。 一旦所有的峰都经过测试,剩下的信号将重新组合以产生最终的多重峰46。图4显示了此建模过程的一个示例。
使用该建模过程,还可以选择性去除溶剂峰和其它污染物。 所使用的溶剂由用户定义以调整DFT溶剂模型。 为了识别实验数据中的溶剂多重峰,对光谱中预期包含该溶剂的每个峰进行打分, 打分值考虑了峰位置和围绕每个峰的振幅与预期溶剂多重峰的模式匹配接近程度以及距预期溶剂位置的距离。从模型中去除与模拟溶剂多重峰最接近的匹配峰,得到最后的光谱(请参见ESI第S2.1.9节)。
最后,必须对1H光谱中的多重峰进行积分。由于氢的1H同位素丰度为100%,因此光谱中多重峰的积分与每个化学环境中的质子数成正比。 如果可以估算该比例常数,则可以明确告诉归属算法(AA)每个多重峰可以分配多少个质子。
程序中用于估算1H谱的比例常数的算法是从这一领域的先前工作发展而来的。本算法的前提是将这个常数k从光谱中最小可能质子数(结构中质子数减去不稳定的质子数)迭代到最大值(设置为总量的两倍) 在模糊结构中的质子数),并根据相应的积分集合的积分计算分数(多重态的积分使用Schoenberger et al.45所述的模型光谱计算)。 最高分的k值作为比例常数,并用于归一化积分(ESI第S2.1.10 †节)。这种打分方法是特别有利的,因为其考虑了由于例如匀场参数的选择或不完全弛豫而经常观察到的与整数积分值的偏差。 使用类似算法执行13 C谱的峰采集。选择光谱中最强的峰,并用Lorentzian函数对其拟合以创建初始模型,然后对下一个最强峰重复此过程。 这个过程持续到所有未捡拾的峰落在拟合模型噪声的标准偏差的三倍内。选择该算法是因为它可以有效地去除噪声峰,同时还能识别低强度信号峰,例如四级碳。
归属算法
开发DP4-AI最后挑战是归属算法(assignment algorithm,AA),其将分子的每个非对映异构体中的原子归属到光谱中观测到的峰,归属依赖于GIAO预测的化学位移而实现。
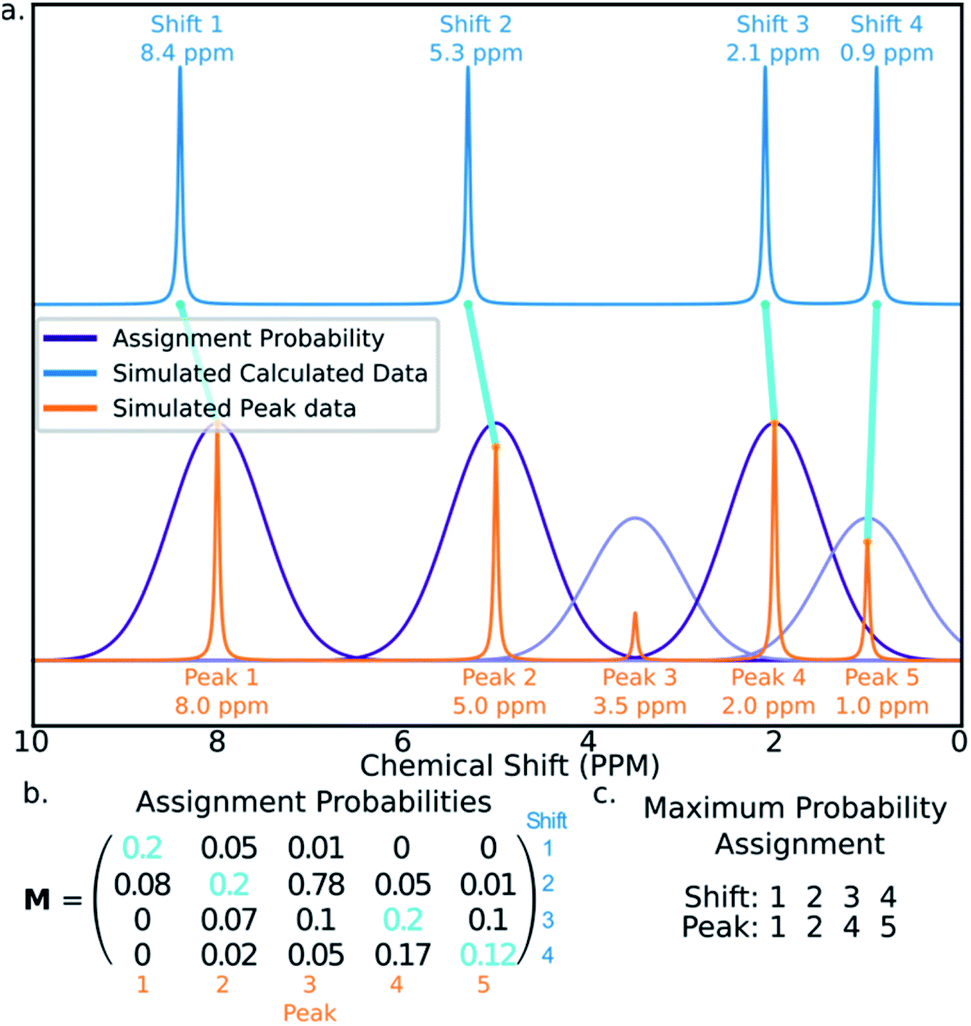
Figure 5. 该图说明了如何使用归属概率矩阵M将计算的位移归属到实验峰上。(a)将模拟计算出的光谱中的峰(蓝色)归属到实验光谱中的峰(橙色)。 (b)计算矩阵M并计算最佳归属(青色)。(c)本算例找到的最终归属。
AA的核心是计算归属概率矩阵M。此矩阵Mij的元素给出计算化学位移i与实验峰j对应的概率。如图5所示,矩阵M用Hungarian线性总和最小化方法找出最可能的归属47。
M值使用统计模型(ESI第S2.2节)来计算,该模型考虑了在所选计算条件下观察到的DFT预测误差的分布,并且在13C-NMR的情况下,还考虑了实验峰的振幅。
GIAO化学位移预测会受到系统误差的影响,系统误差随化学位移在谱图中的位置和计算条件的变化而变化12,48。在计算M之前必须纠正这些系统误差。经典DP4通过内部缩放来解决此问题3。由于归属是未知的,因此无法在此程序中使用该方法。
为了缓解此问题,归属过程分三个阶段执行。在第一轮归属中,在计算M之前,使用已知的外部缩放因子(ESI第S2.2.1†节)执行线性缩放。 在完成第一轮的归属之后,以与DP4类似的方式,使用分归属的化学位移和峰来计算内部线性比例因子。 然后重新对计算的化学位移进行缩放并重新归属。
在13C谱中,实验峰的数量可能不等于分子中的碳原子数。GIAO的化学位移预测也不可能反映光谱中出现的简并。使用Eq(1)的惩罚系统,向13C提供额外的灵活性以多次归属光谱中的峰值。
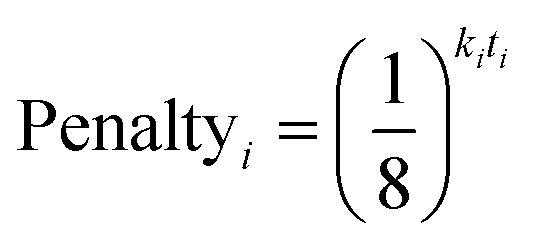 |
(1) |
实验峰i ki的多次归属罚分取决于峰i所在KDE组值。最强峰的组其k = 1的,第二强峰的组k = 2,t的值表示峰已被归属的次数。
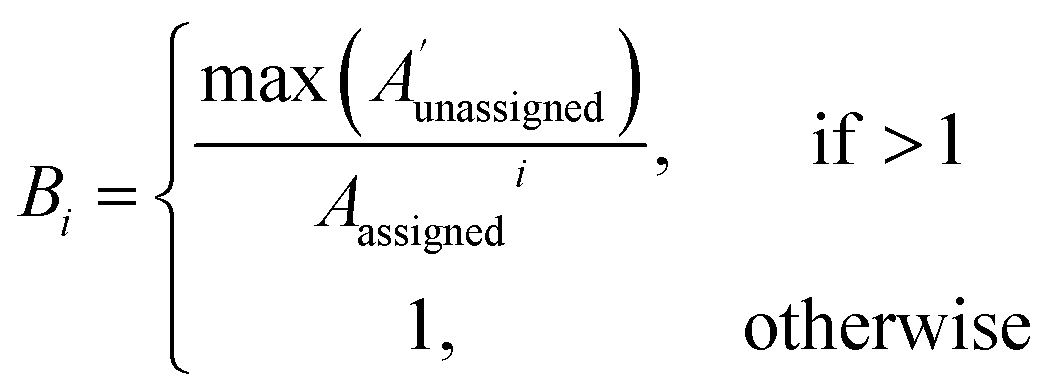 |
(2) |
上面(EQ2)给出了化学位移i的偏差。 其中Aunassigned是一个矢量,其包含了在归属给计算的化学位移i峰的+/- 10 ppm内所有未归属峰的振幅,而Aunassigned i是归属到计算的化学位移i的峰的振幅权重。
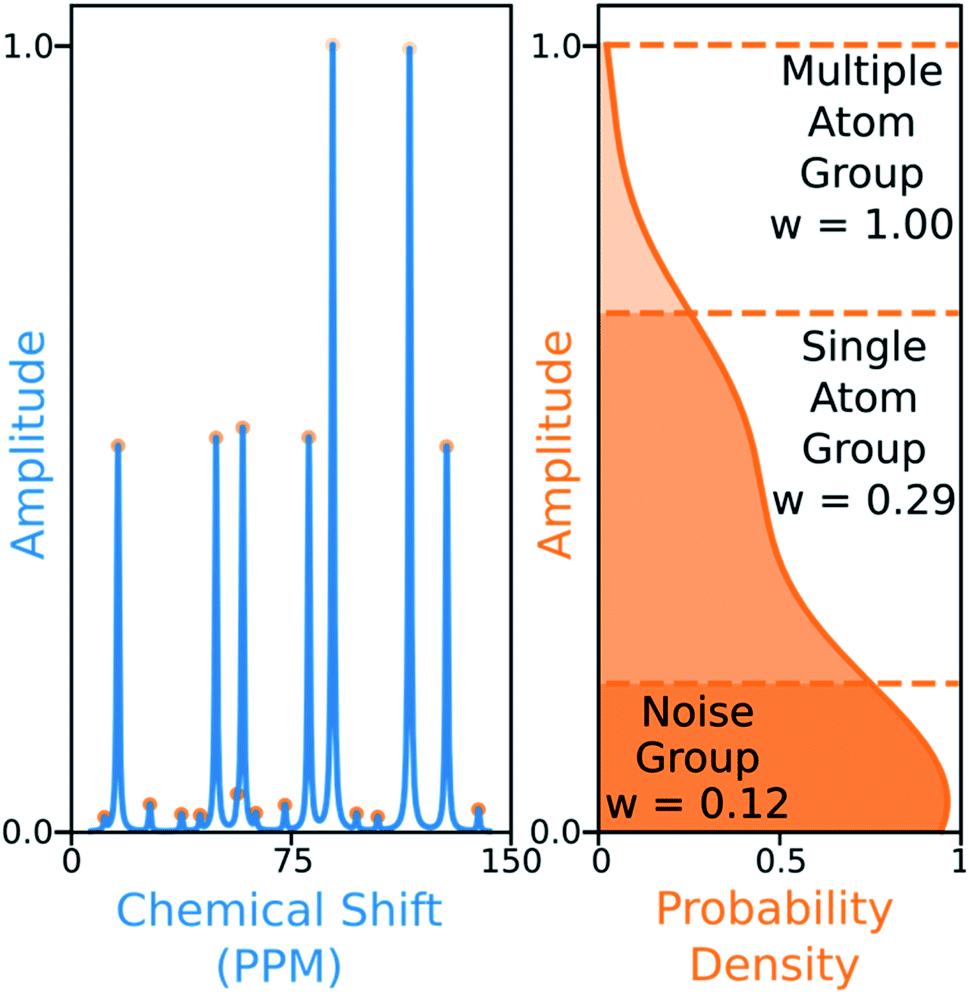
Figure 6. 根据振幅概率密度函数的二阶导数的最小值(右侧),峰(左侧)按振幅分组(落在虚线之间)。 本算例的碳原子数为9。 计算每个组下边界上方的峰的累积总和,分配给每个组的权重是结构中碳原子的数量除以该值。然后将权重标准化以将最大权重固定为1。
13C算法还考虑了实验峰的振幅,将M的每个元素Mij乘以从实验峰j振幅导出的权重Aj,这可以优先归属到那些更强的峰而不是更可能是噪音的峰。13C光谱中的峰通常分为三组,这三组通过幅度来区分:噪声,1-原子信号和对应于多个等价碳原子的信号。为了捕获这种变化,需估算光谱中峰振幅的概率密度函数49。根据该函数的二阶导极小点所处的位置对峰进行分组。然后使用每组中的峰数和结构中碳原子的预期碳原子数来计算振幅权重,如图6所示。
通过考虑峰值强度的分布和每个计算的化学位移周围的局部环境中的位置,13C的归属算法还能够将归属偏向于位置或幅度信息(ESI部分S2.2.2 †)。 在第二轮归属之后,分析归属到每个计算化学位移的实验峰的10ppm范围内的未归属峰。所计算的位移i的偏差由方程(2)给出。在振幅的10ppm范围内,将偏差高于1的所有化学位移按照偏差的顺序重新归属到未归属的实验峰。
偏差的作用是评估初始归属是否错过任何信号峰。这对噪音大的光谱特别有用,因为归属算法通常倾向于在第一轮中归属接近的噪音峰而不是更远的强信号峰。
相反,1H归属算法不需要幅度加权、偏置或多重归属的代价,因为该归属算法可以使用积分信息明确地知道每个峰可以归属多少次。1H归属算法还可以用来归属甲基质子。甲基中的质子在1H-NMR光谱中一致地表现为等价的,因此应该被指认为同一个峰。在对其它质子的归属之前,1H归属算法将这些质子成组地归属到具有足够积分的峰上。
图形用户界面
DP4-AI支持命令行与图形界面的全自动工作流。图形用户界面使用户可以轻松计算DP4概率,可视化DP4-AI归属,并分析构象分布和预测误差。
结果
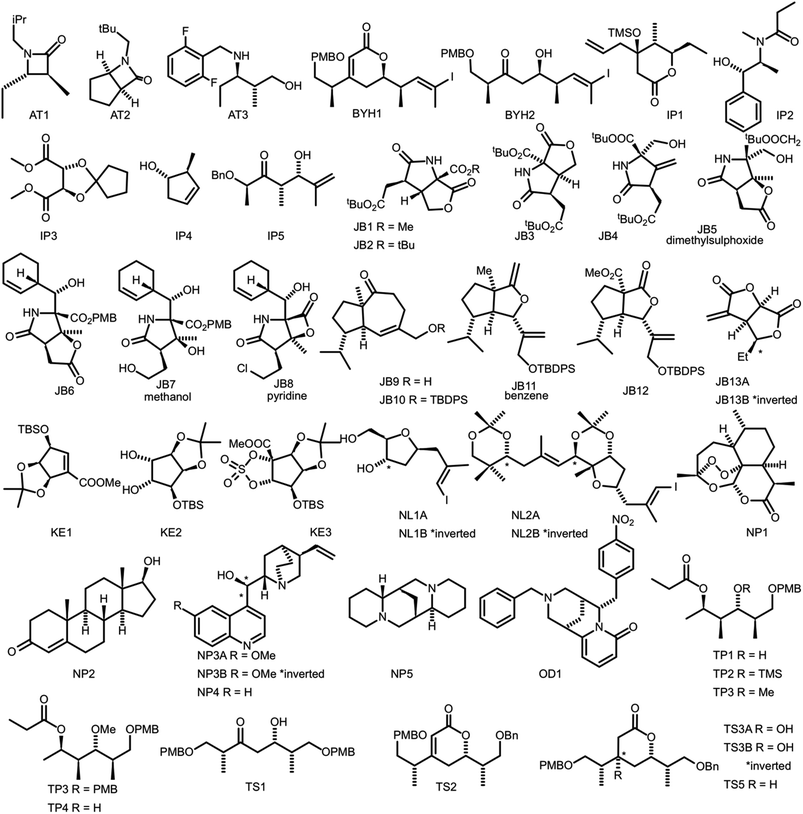
Figure 7. 用来评估DP4-AI性能的47个算例化合物
为了评估NMR-AI的性能,构建了一个包含47个分子(平均每个分子3.49立体化学中心)的测试集,具有各种类型的碳骨架(图7)50-55。该测试集包含了天然产物、合成中间体和天然产物片段,这些化合物代表了DP4-AI可能遇到的使用场景。这些分子对归属算法和DP4均具有挑战性。先前的工作12,13已经证明,柔性的结构,尤其是五元环,以及良好分离的立体化学中心,使得光谱解释变得困难。预期所有这些分子将对DP4-AI提出重大挑战。一套更小、更刚性的分子数据集则更易于分析。还用一系列溶剂中测定了相应的光谱,其中一些显示出非常低的信噪比,而一些为混合物。对该测试集的测试代表了对DP4-AI性能的严格测试。
为了用当前DP4版本预测分子的相对立体化学,用户必须提供NMR描述。在NMR描述中需要的最小信息量是实验峰位、分子中哪些原子化学等价的描述或者每个峰可以被归属的次数。利用该信息,DP4将分子中的原子按照化学位移的顺序归属到NMR描述中的峰。我们将这种方法称为“成对AA(Pairwise AA)”,并将其用作与DP4-AI进行比较的基准。
对测试集中的所有分子采用成对AA方法进行归属,这是一项非常困难的工作,因为需要手动分析所有NMR谱,以便将信号分解成各个峰和多重峰。这是经典DP4最耗时的部分,也具有主观性和引入错误的可能性。 使用三组不同的计算条件来计算DP4概率。 所测试的第一个理论水平是MM获得的几何构型,采用以前工作11建议的mPW1PW 91/6-311G(d)(含碘分子用mPW1PW 91/def2-SVP)和PCM溶剂模型用GIAO预测NMR化学位移。还使用B3LYP泛函在DFT水平优化几何构型后进行了DP4计算。 DFT优化的几何结构,再用M06-2X/def2-TZVP计算单点能量。
DP4还需要统计模型以描述NMR化学位移预测的误差概率。由于预测误差分布预期随计算条件而改变,因此对于所使用的每组条件需要不同的模型。 对四种不同的统计模型进行了测试(ESI部分S3.1 †),发现利用从测试集导出的经验预测误差分布拟合的单域3高斯模型获得了最高的性能。 由于该统计模型是使用测试集中的分子构建的,并且还被用于计算相同测试集的DP4概率,因此还完成了交叉验证研究,以评估是否发生了任何过度拟合。 该交叉验证研究是以图7中所示的各组分子的首字母缩写形式进行的。

Figure 8. DP4-AI与Pairwise AA的正确预测率
在每个统计模型(ESI部分S3 †)描述的三个理论水平上对DP4-AI进行了测试。最高级别理论水平和最可靠的统计模型的DP4-AI和配对AA的比较如图8所示。
讨论
在最高级别的理论水平测试下,DP4-AI解释光谱的可靠性与传统的劳动密集型成对AA相似,后者需要训练有素的化学家对光谱进行预处理(图8)。考虑到数据集的挑战性,这是一个令人印象深刻的结果。 在此数据集中正确有效地归属此立体化学的可能性约为3 x 10-8,这表明DP4-AI的性能非常可靠,远胜于偶然(ESI第S3†节)。 最令人印象深刻的是,DP4-AI从可能的32和64个非对映异构体中正确归属了NP1和NP2的相对立体化学。成对AA代表了本研究中DP4-AIs性能的上限,因为成对AA所使用的NMR描述已被精心编写以消除任何错误。实际上,错误通常发生于NMR描述和归属中,在这种情况下,NMR-AI可能会胜过成对的AA。
比之成对AA,DP4-AI的性能随理论水平级别的提高而提高(图8)。 如先前的工作13所示,随着DP4计算中理论水平级别的提高,成对AA的正确预测率也会提高。DP4-AI对理论水平显示出更大的敏感性。这是因为归属和DP4计算都取决于NMR化学位移计算的精确度。因此,可以得出结论,当使用DP4-AI时,应始终使用产生最准确的化学位移预测条件。

Figure 9. DP4-AI处理、归属BYH1的1H谱(溶剂为氯仿)
通过可靠地解决GIAO NMR预测中仍然存在的挑战,包括构象柔性、特定的溶剂相互作用和重原子的存在,可以进一步改善DP4-AI的性能。通过增加对混合物的光谱(例如IP2,请参见ESI第S3.2节)的显式支持,可以进一步改善性能。这些问题将在DP4-AI后续的开发中解决。图9给出了一个由DP4-AI归属的光谱算例(所有处理和归属的光谱均可参见ESI的S4†部分)。
结论
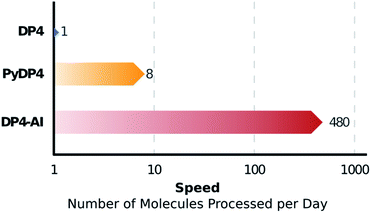
Figure 10. NMR-AI在1分钟内处理完用于DP4计算的分子,而在此之前大约需要8个小时的用户时间。这相当于每天可处理的分子数量增加约60倍。
DP4-AI是一个稳健的系统,可自动处理和归属原始的13C和1H-NMR光谱,自动解释结构不确定性,并已作为开放源码软件发布。其自动化使得对数据库和大量分子进行快速DP4分析成为可能(图10)。比之利用专业化学家编写NMR描述的DP4,DP4-AI具有同样的正确结构确证率。此外,该系统能够可靠地处理和归属NMR光谱,但速度快约60倍,从而释放了化学家用于实验和发现的时间。此外,该新系统为将来开发新功能(例如J值分析,2D NMR归属,归属复杂混合物的光谱和辅助构象分析)提供了鲁棒的框架。DP4-AI作为开源软件,可从网址https://github.com/KristapsE/DP 4-AI获得。
DP4-AI源代码下载
https://github.com/KristapsE/DP4-AI
DP4-AI的使用
请参见:DP4-AI教程 | 自动DP4计算。
手工计算DP4概率
如果你有实验NMR数据,你也计算了各个异构体的NMR,然后可以根据Smith2010的方法计算DP4概率,具体操作步骤见:Goodman课题组网站或博文DP4概率的计算。当DP4-AI的归属出现错误时,可以利用DP4-AI的计算NMR与实验NMR进行手动DP4计算。
Smith, S. G.; Goodman, J. M. Assigning Stereochemistry to Single Diastereoisomers by GIAO NMR Calculation: The DP4 Probability. J. Am. Chem. Soc. 2010, 132 (37), 12946–12959. https://doi.org/10.1021/ja105035r.


