
摘要:本文以化合物IP4为例,演示了如何用DP4-AI的工作流自动进行立体化学结构确证。
作者:肖高铿
日期:2020-10-18
关于DP4-AI
在前文DP4-AI自动NMR数据分析:直接从光谱到结构中,我们详细介绍了DP4-AI。虽然有AI字样,但与目前火热的AI概念并无任何关系。相反它是基于物理的方法进行NMR预测,然后通过比较理论NMR与实验NMR来识别可能的差向异构体。假设你有NMR原始数据(13C-NMR和/或1H-NMR数据),你可以通过DP4概率的计算来识别您的化合物可能是差向异构体中的哪一个。本文将通过一个算例演示如何使用DP4-AI进行全自动的DP4计算。
DP4-AI下载
下载:https://github.com/KristapsE/DP4-AI
1 | git clone https://github.com/KristapsE/DP4-AI.git |
算例介绍
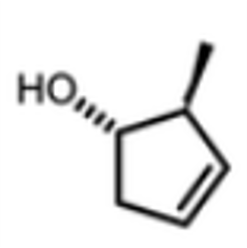
Figure 1. 算例化合物IP4
本算例化合物IP4的结构如图1所示,已经准备好了3D的SDF文件、13C-NMR和/或1H-NMR原始数据(下载),这些文件保存在同一目录下,结构如下:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 | demo ├── IP4_.sdf └── NMR_folder ├── Carbon │ ├── acqu │ ├── acqus │ ├── audita.txt │ ├── cpdprg2 │ ├── fid │ ├── format.temp │ ├── fq1list │ ├── pdata │ │ └── 1 │ │ ├── 1i │ │ ├── 1r │ │ ├── auditp.txt │ │ ├── outd │ │ ├── proc │ │ ├── procs │ │ ├── thumb.png │ │ └── title │ ├── pulseprogram │ ├── scon2 │ ├── shimvalues │ ├── specpar │ ├── uxnmr.info │ └── uxnmr.par └── Proton ├── acqu ├── acqus ├── audita.txt ├── fid ├── format.temp ├── fq1list ├── pdata │ └── 1 │ ├── 1i │ ├── 1r │ ├── auditp.txt │ ├── outd │ ├── proc │ ├── procs │ ├── thumb.png │ └── title ├── pulseprogram ├── scon2 ├── shimvalues ├── specpar ├── uxnmr.info └── uxnmr.par 7 directories, 42 files |
你需要下载该文件到您的一个目录下,解压,并切换到该目录。到此,你已经准备好开始DP4-AI计算了。
DP4-AI的使用
1.使用方法
键入PyDP4 -h命令获得帮助:
1 | python /<安装路径>/DP4-AI/PyDP4.py -h |
其中“安装路径”用你实际的路径代替,键入命令后出现帮助提示,按提示完成指令键入。
2. 起始的文件
以作者用到化合物IP4为例,以说明DP4计算的流程。初始的输入信息包括:
- IP4的3D结构
- NMR实验数据
IP4的3D结构以MDL SDF格式保存为IP4_.sdf
原始的13与C1H-NMR实验数据分别以Carbon与Proton命名,保存在目录NMR_Folder里。
3. 使用gmns工作流
现在,可以开始用DP4-AI的自动工作流开始进行DP4计算。我们先试试作者的MM方法(直接用分子力场的构象进行NMR计算)。该策略使用工作流(-w gmns)来实现,包含了如下计算过程:差向异构体的枚举(g)、MMFF力场构象搜索(m)、NMR核磁计算、计算的NMR与实验NMR的数据提取与DP4计算。命令如下:
1 | PyDP4.py -s chloroform -w gmns -m t -f mmff IP4_.sdf NMR_folder |
其中-s chloroform是告诉DP4-AI在进行NMR计算时需要考虑溶剂(氯仿)的效应;-m t的意思是用Tinker进行构象搜索。
结果如下:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 100 101 102 103 104 105 106 107 108 109 110 111 112 113 114 115 116 117 118 119 120 121 122 123 124 125 126 127 128 129 130 131 132 133 134 135 136 137 138 139 140 141 142 143 144 145 146 147 148 149 150 151 152 153 154 155 156 157 158 159 160 161 162 163 164 165 166 167 168 169 170 171 172 173 174 175 176 177 178 179 180 181 182 183 184 185 186 187 188 189 190 | ========================== PyDP4 script, integrating Tinker/MacroModel, Gaussian/NWChem and DP4 v1.0 Copyright (c) 2015-2019 Kristaps Ermanis, Alexander Howarth, Jonathan M. Goodman Distributed under MIT license ========================== Settings read from settings.cfg: TinkerPath: /home/cloudam/software/tinker NWChemPath: /public/software/.local/easybuild/software/NWChem/7.0.0-intel-2019b-Python-3.7.4/bin/nwchem GausPath: g16 ['IP4_.sdf'] NMR_folder NMR_path /home/cloudam/work/dp4/IP4_tinker/NMR_folder Current working directory: /home/cloudam/work/dp4/IP4_tinker Initial input files: ['IP4_.sdf'] NMR file: [PosixPath('/home/cloudam/work/dp4/IP4_tinker/NMR_folder/Carbon'), PosixPath('/home/cloudam/work/dp4/IP4_tinker/NMR_folder/Proton')] Workflow: gmns Generating diastereomers... Getting inchi from file IP4_.sdf Getting inchi from file IP4_.sdf Number of diastereomers to be generated: 2 Isomer 0 inchi = InChI=1S/C6H10O/c1-5-3-2-4-6(5)7/h2-3,5-7H,4H2,1H3/t5-,6-/m0/s1/f/ Isomer 1 inchi = InChI=1S/C6H10O/c1-5-3-2-4-6(5)7/h2-3,5-7H,4H2,1H3/t5-,6+/m0/s1/f/ Generated input files: ['IP4_1', 'IP4_2'] Assuming all computations are done? ... False Using preexisting DFT data? ... False Performing conformational search using Tinker Setting up Tinker files... Tinker input for IP4_1 prepared. Tinker input for IP4_2 prepared. Running Tinker... Output files for IP4_1 already exist, skipping. Output files for IP4_2 already exist, skipping. IP4_1 is matching conformational search output for IP4_1 Reading IP4_1 Number of accepted conformers by energies: 3 IP4_2 IP4_1 Nope IP4_1 IP4_2 Nope IP4_2 is matching conformational search output for IP4_2 Reading IP4_2 Number of accepted conformers by energies: 5 Pruning conformers... IP4_1: 3 conformers after pruning with 0A RMSD cutoff IP4_2: 5 conformers after pruning with 0A RMSD cutoff Setting up NMR calculations... Running NMR calculations... Running Gaussian DFT NMR calculations locally... There were no jobs to run. Reading data from the output files... IP4_1ginp001.out 17 IP4_1ginp002.out 17 IP4_1ginp003.out 17 IP4_2ginp001.out 17 IP4_2ginp002.out 17 IP4_2ginp003.out 17 IP4_2ginp004.out 17 IP4_2ginp005.out 17 Shieldings: IP4_1: [52.7725, 142.6171, 106.6767, 137.7624, 44.3868, 262.8271, 169.0847, 29.3631, 29.358, 30.1239, 27.8744, 26.3857, 26.1914, 31.7494, 31.5088, 30.8564, 30.9325] [51.1922, 147.0432, 106.3846, 133.0215, 45.7752, 264.1163, 169.3307, 29.4695, 29.7625, 29.6988, 27.8628, 26.3143, 26.2597, 31.6062, 31.5343, 30.9341, 31.0055] [52.1369, 144.3506, 107.3824, 134.4598, 45.1125, 256.7895, 169.721, 29.3409, 29.4651, 29.8111, 28.2077, 26.3773, 26.2689, 30.9988, 31.5238, 30.8044, 30.9704] IP4_2: [52.5388, 147.5788, 111.0206, 142.2785, 45.1509, 290.3178, 172.6921, 29.6835, 29.5192, 29.4242, 27.249, 26.4213, 26.3882, 31.3379, 31.1798, 31.191, 31.1351] [53.7533, 143.4332, 112.0511, 142.9355, 44.3782, 280.9794, 172.8862, 29.8359, 29.2278, 29.4755, 27.2404, 26.4982, 26.3565, 31.3338, 31.2015, 31.1403, 30.7387] [51.0884, 141.8932, 111.8456, 140.8611, 45.4508, 282.0051, 173.2361, 29.7459, 29.3937, 29.3827, 28.0109, 26.3166, 26.4308, 31.3822, 30.8604, 31.2061, 30.909] [50.699, 144.5118, 112.3287, 140.6304, 45.8268, 292.835, 175.1699, 29.5901, 29.4615, 29.4456, 27.7753, 26.259, 26.4269, 31.5163, 30.8012, 31.0402, 31.2906] [52.0486, 140.3173, 112.6003, 140.7325, 44.5733, 275.5862, 174.1799, 30.0199, 29.3096, 29.4031, 27.7075, 26.3135, 26.3433, 31.8847, 30.9336, 30.8427, 30.8827] Energies: IP4_1: [-309.84674683, -309.847000007, -309.847961686] IP4_2: [-309.846569715, -309.845282065, -309.847936164, -309.847324941, -309.845372646] Setting TMS computational NMR shielding constant references Setting TMS references to 188.452125 and 32.1243166667 Converting DFT data to NMR shifts... [0.16868446982677085, 0.22056053309520496, 0.6107549970780243] [0.12478688500156183, 0.03190761049375939, 0.5305037184618006, 0.277681469399478, 0.03512031664340019] WARNING: NMR shift calculation currently ignores the instruction to exclude atoms from analysis C shifts for isomer 0: 136.442, 43.808, 81.424, 53.763, 143.343, 18.928 H shifts for isomer 0: 2.751, 2.612, 2.285, 4.049, 5.760, 5.871, 0.865, 0.601, 1.283, 1.153 C shifts for isomer 1: 137.198, 45.137, 76.557, 47.425, 143.026, 14.728 H shifts for isomer 1: 2.417, 2.704, 2.715, 4.309, 5.805, 5.706, 0.694, 1.227, 0.981, 1.088 Reading experimental NMR data... [PosixPath('/home/cloudam/work/dp4/IP4_tinker/NMR_folder/Carbon'), PosixPath('/home/cloudam/work/dp4/IP4_tinker/NMR_folder/Proton')] Cshifts [16877, 24398, 27885, 35275, 36948, 41526, 44704, 45401, 48935, 49415, 49494, 50315, 51088, 51312, 52165, 52748, 52866, 52924, 52967, 53041, 53446, 53492, 53797, 53892, 54317, 54566, 56173, 56629, 56817, 56866, 56916, 57006, 57260, 57585, 57629, 59516, 60025, 62380, 74728, 76429, 76985, 78309, 80656, 80954, 81834, 81886, 81995, 82055, 82156, 82247, 82804, 83202, 83742, 83834, 84531, 84971, 88667, 88872, 92159, 94973, 96204, 96290, 96392, 97478, 98130, 98381, 98466, 98885, 99220, 99411, 99539, 99647, 99667, 99746, 100137, 100234, 100334, 100702, 100929, 101103, 101319, 101365, 101402, 101596, 102180, 103160, 104388, 105039, 108737, 109250, 110025, 110449, 111396, 111600, 111829, 111913, 112591, 112813, 113438, 113480, 113943, 114030, 114676, 115240, 127011] Assigning carbon spectrum... Plotting carbon spectrum... Assigning proton spectrum... Plotting proton spectrum... Calculating DP4 probabilities... none No stats model provided, using default number of c protons = 10 number of c carbons = 6 number of e protons = 10 number of e carbons = 6 IP4_1 Solvent = chloroform Force Field = mmff DFT NMR Functional = mPW1PW91 DFT NMR Basis = 6-311g(d) Number of isomers = 2 Number of conformers for isomer 1 = 3 Number of conformers for isomer 2 = 5 Assigned C shifts for isomer 1: label, calc, corrected, exp, error C7 18.93 15.82 16.23 0.41 C2 43.81 39.16 39.34 0.18 C4 53.76 48.50 47.15 -1.35 C3 81.42 74.44 75.43 0.99 C1 136.44 126.05 125.26 -0.79 C5 143.34 132.52 133.08 0.56 Assigned C shifts for isomer 2: label, calc, corrected, exp, error C7 14.73 15.72 16.23 0.51 C2 45.14 43.41 39.34 -4.07 C4 47.43 45.50 47.15 1.65 C3 76.56 72.03 75.43 3.40 C1 137.20 127.26 125.26 -2.00 C5 143.03 132.57 133.08 0.51 Assigned H shifts for isomer 1: label, calc, corrected, exp, error H15 0.60 0.71 0.95 0.24 H14 0.86 0.95 1.66 0.71 H17 1.15 1.22 0.95 -0.27 H16 1.28 1.34 0.95 -0.39 H10 2.29 2.27 2.18 -0.10 H9 2.61 2.58 2.55 -0.02 H8 2.75 2.71 2.55 -0.15 H11 4.05 3.91 3.91 -0.01 H12 5.76 5.50 5.54 0.04 H13 5.87 5.61 5.54 -0.06 Assigned H shifts for isomer 2: label, calc, corrected, exp, error H14 0.69 0.73 1.66 0.93 H16 0.98 1.00 0.95 -0.05 H17 1.09 1.10 0.95 -0.15 H15 1.23 1.23 0.95 -0.28 H8 2.42 2.35 2.18 -0.17 H9 2.70 2.62 2.55 -0.07 H10 2.72 2.63 2.55 -0.08 H11 4.31 4.13 3.91 -0.23 H13 5.71 5.45 5.54 0.10 H12 5.81 5.54 5.54 0.00 Results of DP4 using Proton: Isomer 1: 97.2% Isomer 2: 2.8% Results of DP4 using Carbon: Isomer 1: 99.3% Isomer 2: 0.7% Results of DP4: Isomer 1: 100.0% Isomer 2: 0.0% PyDP4 process completed successfully. |
可以看到,本次DP4-AI工作流对化合物进行了差向异构体枚举,得到两个结构isomer 1与Isomer 2; 然后分别对两个异构体进行构象搜索,异构体1、2分别得到3与5个构象;分别对每个构象进行NMR计算、根据构象能得到计算的NMR数据;并提取实验NMR数据;然后进行DP4计算,分别给出了NMR实验数据归属为两个异构体的DP4概率。
最重要的是,在归属的过程中,我们没有事先对1H与13C-NMR实验数据进行预处理与归属,这全部由DP4-AI自动动进行。比起需要花大量的时间编写NMR描述数据的常规方法DP4计算,DP4-AI的自动NMR数据处理为化学科研人员节省了大量的宝贵时间。
除了给出DP4概率之外,计算与实验图谱的归属也生成在一个新的目录Graphs里,每个异构体分别一张13C与1H-NMR图谱, 以矢量图方式给出。图11与12是其中一个异构体的13C与1H-NMR图谱。

Figure 11. 13C-NMR实验与计算图谱及其归属(矢量图,可缩放)

Figure 11. 1H-NMR实验与计算图谱及其归属(矢量图,可缩放)
积分也是自动完成,生成的矢量图里已经指认好了计算峰与实验峰,通过颜色进行对映,溶剂峰的着色也区别其它峰。结合给出的各个异构体的SDF文件,可以很方便的将化合物编号与图谱编号进行比对解读。
除了给出图形格式的NMR数据,还新生成了一个目录Pickles, 该目录将将实验NMR数据以数据文件的格式保存,可以用Pickle模块读取。此外,还新生成文件ANPN-D-4-60_1NMR.dp4,包含了dp4计算过程的信息信息。新生成的两个目录与一个文件结构如下:
1 2 3 4 5 6 7 8 9 10 11 | Graphs
└── IP4_1
├── Carbon_1.svg
├── Carbon_2.svg
├── Proton_1.svg
└── Proton_2.svg
Pickles
└── IP4_1
├── carbondata
└── protondata
IP4_1NMR.dp4 |
注意:如果你有第二个异构体的NMR数据,重新进行DP4计算此时,需要将Graphs与Pickles更名再计算DP4,并且IP4_1NMR.dp4会被重写。
手工计算
根据博文《DP4概率的计算》所描述的方法,Isomer_1/2的学生氏t-分布与Gaussin概率分布计算的DP4概率分别为98.97%/99.46%与1.03%/0.44%。这与DP4-AI自动计算的DP4概率99.3%与0.7%基本一致。
注意事项
DP4-AI自动读取实验数据并进行归属与积分并非万无一失。实践中发现,很多因素会导致自动积分错误与归属错误。比如有杂质峰的时候会出现积分错误和/或归属错误,虽然此类错误在化学工作者看来显而易见。因此,强烈建议:在采用DP4之前,对生成的图谱与归属进行复核。
DP4计算的布署与培训
部署与培训服务,请联系我们。


