
摘要:人工智能(AI)在药物发现中的应用已成为近年来研究的热点。基于深度学习的生成分子设计是个特别值得关注的领域。 Zavoronkov等人最近发表了一种新方法,使用了基于深度学习的从头分子设计用于发现新型、有效的DDR1激酶抑制剂。从模型建立到化合物设计花了21天,总共合成并测试了6个AI设计的化合物。这项研究强调了AI设计化合物领域发展的速度有多快,我们可以预期在未来几年会有进一步的发展。
编译:肖高铿/2019-12-15
原文:Chen H, Engkvist O. Has Drug Design Augmented by Artificial Intelligence Become a Reality? Trends Pharmacol Sci. October 2019:9-11. doi:10.1016/j.tips.2019.09.004
人工智能(AI)在药物发现中的应用已成为近年来研究的热点。基于深度学习的生成分子设计是个特别值得关注的领域。 Zavoronkov等人最近发表了一种新方法,使用了基于深度学习的从头分子设计用于发现新型、有效的DDR1激酶抑制剂。从模型建立到化合物设计花了21天,总共合成并测试了6个AI设计的化合物。这项研究强调了AI设计化合物领域发展的速度有多快,我们可以预期在未来几年会有进一步的发展。
药物发现以其漫长且昂贵的研发过程而闻名,通常被形容为从干草堆中寻找一个针头。 在这种情况下,这个干草堆包含大约1060-10100个可合成的分子[1],我们需要从中找到一个完美的化合物,同时满足包括生物活性、药物代谢和药代动力学( DMPK)性质特征、合成可行性等多种标准。从药物研发立项开始到临床前研究候选药物的确定过程通常需要3-5年,并且需要合成和测试数百至数千种化合物。开发从头设计算法来虚拟设计、评估化合物可以减少寻找合适针头的时间与成本,尤其是将AI设计与自动化化学相结合的话。
早期的从头设计算法[1]使用基于结构的方法来生长出在空间上和静电上都与靶标结合口袋匹配的配体。这种方法设计的化合物通常DMPK特性不好并且化学上难以合成[2]。另一种从头设计的方法是首先枚举大型虚拟库,然后用分子对接、相似性搜索、药效团搜索来探索化学空间[3]。库的枚举通常基于预先定以的反应和市售试剂。另一种方法是根据药物化学家的经验[4],使用转化规则对查询结构(query structure)进行增量修饰。所有这些方法的共同问题是预定义的反应/转换规则的固有严格性和使用范围。随着深度学习方法的发展,生成模型已是一种令人感兴趣的从头设计方法[5]。通过对大量化学结构概率分布的学习,生成模型可以学习到类药化合物应该的样子,而无需引入任何严格的规则。递归神经网络(RNN)[6,7],变分自编码器(VAE)[8]和对抗生成网络(GAN)[9]是生成模型方法中最常用的深度学习架构。 Merket等人报道了使用生成模型进行分子设计并进行实验验证的第一个例子。其中RNN模型用来设计视黄醇X(Retinoid X)和PPAR受体激动剂。设计的化合物在基于细胞的活性测试中显示出纳摩尔至低微摩尔的受体调节活性[10]。
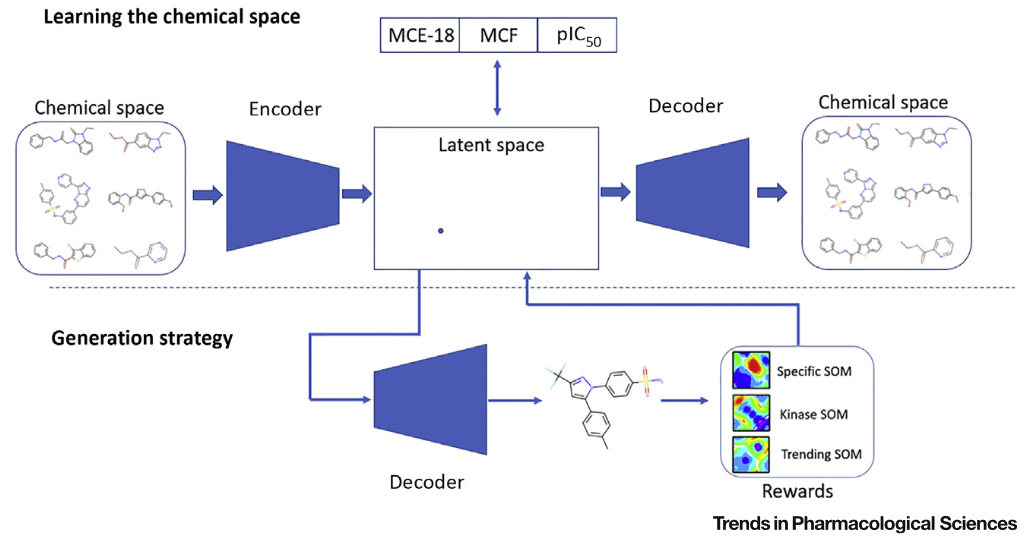
Figure 1. GENTRL分子设计工作流
最近,Zhavoronkov及其同事[11]应用了一种生成张量强化学习(GENTRL)模型(一种深度学习方法)来设计DDR1激酶抑制剂。 GENTRL可以看作是VAE [12]和GAN [9]的进一步发展。他们首先准备了三组化合物数据集用于模型训练:一组是来自ZINC的类先导化合物和代表一般化学空间的内部化合物数据库,第二组是来自Integrity和ChEMBL数据库的激酶抑制剂和非活性化合物,第三组是来自制药公司的专利化合物。基于这些数据集,构建了三个组成GENTRL模型打分函数的自组织图(SOM)[13]模型,以分别评估一般激酶活性,DDR1活性和结构新颖性。 GENTRL模型的核心部分基本上是VAE模型的变体,如图1所示。它采用门控循环单元(GRU;请参阅词汇表)RNN作为编码器,将分子结构(以SMILES字符串表示)映射为潜向量;然后,对潜向量进行解码、重建为输入结构,并且潜向量也同时遵循可学习的先验分布模型。在VAE训练期间,还考虑了各种化合物性质,以便在分子结构、化合物性质和潜向量之间建立映射。在下一步(生成策略,图1)中,进行了强化学习、对模型调参,用奖励函数(特定SOM,一般激酶SOM和趋势SOM,如前所述)偏向性地生成DDR1激酶抑制剂。
作者最初用强化学习GENTRL模型生成了30000个DDR1抑制剂结构。然后用药物化学过滤器、新颖性过滤器、结构聚类,DDR1药效团模型和Sammon映射等级联过滤之后,选择了40个结构,这些结构可以平滑地覆盖最终的化学空间和药效团模型的均方根偏差(RMSD)分布。最终根据合成可行性选择了六个结构进行实验验证。六个化合物均被成功合成,并用酶学激酶测定法测试了体外抑制活性。有两个化合物的DDR1抑制剂活性(IC50)分别为10和21nM,两个化合物具有中等活性,两个化合物没有活性。两个最有效的化合物在随后的细胞活性测试中也显示出强大的活性。在各种DMPK分析中显示出与参照化合物相似的DMPK性质。最后,化合物1以静脉和口服给药方式对小鼠进行测试,并获得了合理的药代动力学结果。化合物1(10mM)针对多个激酶进行了激酶谱测试,仅对DDR1和DDR2显示出大于50%的抑制率。化合物1(最有效的化合物)与Ponatinib(一种用于癌症治疗的激酶抑制剂)具有相同的铰链结合模式,这并不令人惊讶,因为神经网络是用激酶抑制剂进行训练的。
从最初的靶标提名到获得六个化合物的生物学测试结果,整个过程耗时46天。这篇文章在新闻和社交媒体v-vii中引起了极大的关注。这是第一篇包含分子结构及大量实验验证的从头设计范例。该研究强调需要进行广泛的预处理以生成必要的打分模型,以及对从头生成的分子进行后处理,并强调在开发模型和优先考虑合成化合物时必须考虑所有可用信息的重要性。 因此,基于AI的从头设计绝不是一种按一下按钮就完事的活。
这项研究打破了AI生成的化合物难以合成的以讹传讹:本研究的候选化合物可以快速合成。重要的是,作者公开了他们的源代码——应该要承认他们研究的透明性和可重复性已获得支持。然而,重要的是要注意到:本研究的激酶选择性仅覆盖约10%激酶谱,并且就激酶亚家族而言选择不均。此外,尚没有在新合成化合物的活性测试中使用现有的DDR1抑制剂进行基准测试。 尽管这项研究着重介绍了如何将从头设计应用于已有大量信息可供使用的成熟靶标,但人们对诸如激酶和GPCR等被充分利用的靶标之外的全新靶标更感兴趣,希望看到针对全新靶标工作流的样子。
尽管化合物在第一轮设计中已经具有良好的性质,但为了进一步优化化合物1的药效、DMPK分布和激酶选择性以获得临床候选物,更多的设计循环肯定是必要的。然而,作为概念验证,该示例清楚地突显了基于深度学习的生成模型在识别新的先导化合时在时间和成本节约方面对药物发现的潜在影响。生成模型仍处于起步阶段,基于深度学习的从头分子生成仍有进一步改进的空间。我们希望将来会出现更多基于AI的药物设计实例。特别是,我们期待着AI设计的化合物,正如本文所举例说明的,如何与自动化化学相结合使用,以加速新型先导化合物的识别。
文献
- Schneider, G. and Fechner, U. (2005)Computer-based de novo design of drug like molecules. Nat. Rev. Drug. Discov.4,649–6632.
- Schneider, G. and Clark, D.E. (2019)Automated de novo drug design: are we nearly there yet? Angewandte Chemie58,10792–108033.
- Schneider, G.et al.(2011) Reaction-driven de novo design, synthesis and testing of potential type II kinase inhibitors. Future Med. Chem.3,415–4244.
- Besnard, J.et al.(2012) Automated design of ligands to polypharmacological profiles. Nature 492, 215–2205.
- Chen, H.et al.(2018) The rise of deep learning in drug discovery. Drug Discov.Today23, 1241–12506.
- Olivecrona, M.et al.(2017) Molecular de-novo design through deep reinforcement learning. J. Cheminform.9, 487.
- Segler, M.H.S.et al.(2018) Generating focused molecule libraries for drug discovery with recurrent neural networks.ACS Cent. Sci.4, 120–1318.
- Go ́mez-Bombarelli, R.et al.(2018)Automatic chemical design using a data-driven continuous representation of molecules. ACS Cent. Sci.4, 268–2769.
- Putin, E.et al.(2018) Reinforced adversarial neural computer for de novo molecular design. J. Chem. Inf. Model.58, 1194–120410.
- Merk, D.et al.(2018)De novo design of bioactive small molecules by artificial intelligence. Mol. Inform.37,170015311.
- Zhavoronkov, A.et al.(2019) Deep learning enables rapid identification of potent DDR1 kinase inhibitors. Nat. Biotechnol.37, 1038–104012.
- Polykovskiy, D.et al.(2018) Entangled conditional adversarial autoencoder for de novo drug discovery. Mol. Pharm.15, 4398–440513.
- Kohonen, T. (1982) Self-organized formation of topologically correct feature maps. Biol.Cybernetics43, 59–69


